
Abstract
The world is facing a significant environmental crisis as greenhouse gas emissions are negatively impacting the environment, primarily caused by the use of fossil fuels for energy applications. In response, there is a growing effort to develop and adopt sustainable technologies to reduce fossil fuel consumption and to mitigate its harmful environmental effects. Hydrogen fuel cells (HFCs) have emerged as promising alternative energy technologies due to their potential to reduce reliance on fossil fuels and lower greenhouse gas emissions. These technologies utilize platinum group metals (PGMs) as electrocatalysts, due to their exceptional catalytic properties. These catalysts enable high efficiency in hydrogen production and energy conversion. As a result, the global demand for PGMs has increased significantly due to their critical role in modern industrial applications. Given their scarcity and high economic value, the recovery and recycling of PGMs from secondary sources, such as end-of-life materials, has become crucial in advancing these technologies. This paper provides a comprehensive overview of existing trends and advancements in PGM recovery, focusing on the effect of various electrochemical parameters on the dissolution of Pt and Ir electrocatalysts. While conventional recovery methods like hydrometallurgy are effective, they often pose environmental risks due to the release of hazardous effluents. In contrast, electrochemical methods offer a more sustainable alternative for recovering precious metals from spent HFCs, using less harmful processes such as cyclic voltammetry and square wave voltammetry. The research findings highlight the optimal conditions that maximize the recovery of Pt and Ir, providing valuable insights into improving sustainable recovery methods for these critical metals.
This is a visual representation of the abstract.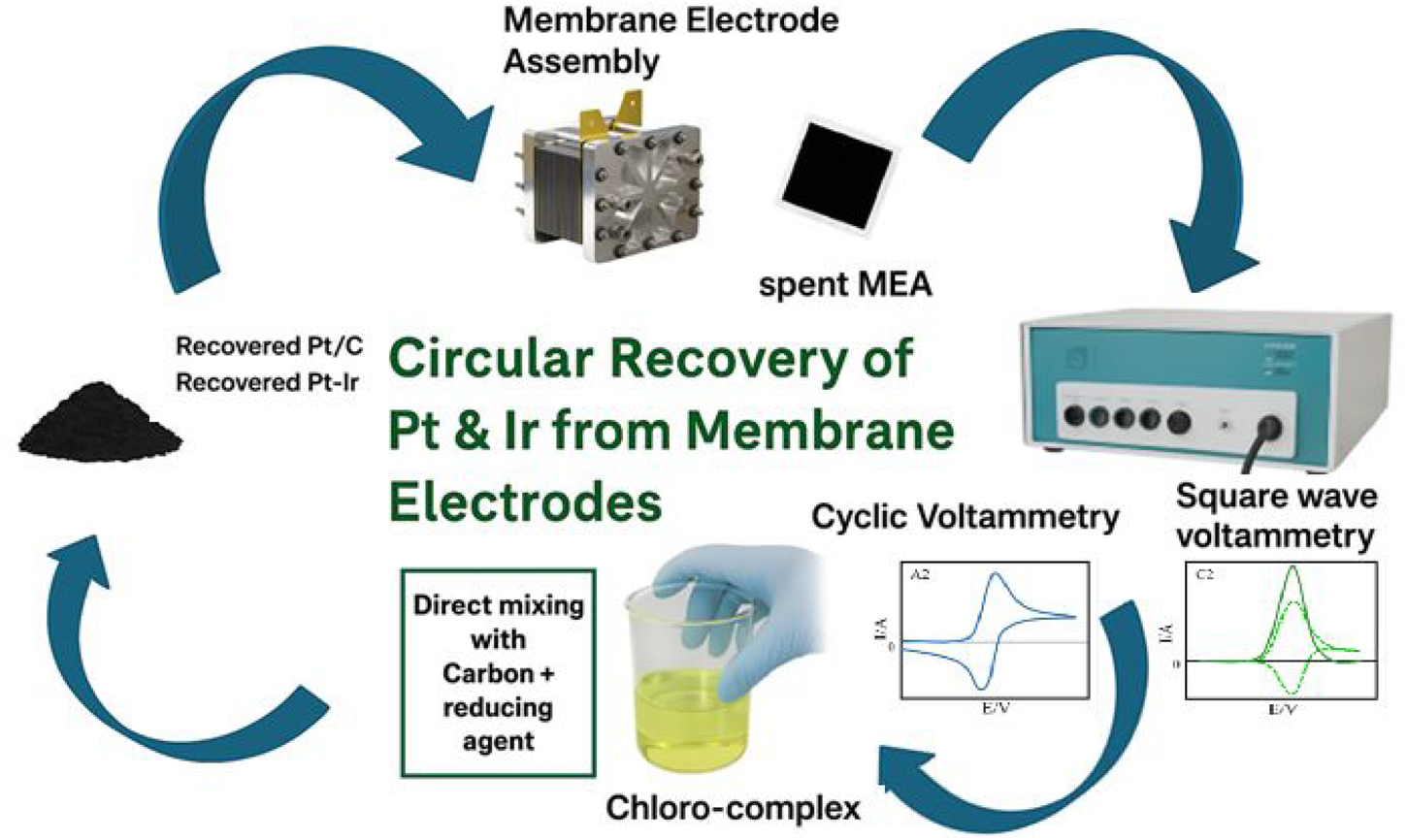
Keywords
Introduction
The need for sustainable energy solutions
The growing global concerns over climate change and the degradation of natural ecosystems due to human activities have placed greenhouse gas (GHG) emissions at the center of the worldwide policy discussions. The primary source of GHG emissions is the combustion of fossil fuels for energy production, industrial processes, and transportation.1,2 The world's transportation usage has doubled in the past 30 years, leading to increased GHG emissions from both fuel production and the transport industry.3,4 As an effort to reduce GHG emissions, hydrogen fuel cells have gained significant momentum as a viable solution, contributing to the prevention of GHG emissions and supporting the European Union's climate action roadmap, which targets an 80% reduction in GHG emissions by the year 2050.5–8 Hydrogen technology offers a promising solution to the global need for clean and sustainable energy, enabling efficient energy conversion with zero emissions while providing additional advantages such as compact design, silent operation, and significantly lower environmental impacts compared to conventional technologies, particularly during operation.5,6,8 The development of hydrogen technologies also plays a crucial role in driving innovation and creating economic opportunities, as their application in the transportation and energy storage continues to expand.9,10
Fuel cells and electrolyzers are rapidly emerging as crucial hydrogen technologies to meet the growing global demand for renewable energy and sustainable fuel alternatives. These systems play a crucial role in reducing reliance on fossil fuels and in lowering GHG emissions by enabling cleaner energy production and storage.11,12 Among the various types of fuel cells and electrolyzers, proton exchange membrane (PEM) systems stand out due to their high efficiency, rapid response time, and adaptability to varying operational conditions.13–15 PEM-based electrochemical systems can operate in both fuel cell and water electrolyzer modes, enabling efficient hydrogen energy utilization and sustainable green hydrogen production. 16 Moreover, PEM water electrolyzers (PEMWEs) and PEM fuel cells (PEMFCs) can be integrated into a single system, forming a closed loop for clean energy storage and conversion. PEM electrolyzers convert electrical energy into hydrogen fuel through electrolysis, allowing energy to be stored as hydrogen. This stored hydrogen can be subsequently used in hydrogen fuel cells to generate electricity, producing water as the only byproduct.12,17,18 This integrated approach not only enhances the efficiency and reliability of renewable energy systems but also paves the way for scalable, sustainable solutions in the transition to a low-carbon economy.
PEM electrolysis is considered a highly effective method for high-purity hydrogen production, due to its high current density (1–3 A/cm2), greater energy efficiency (∼80–86%), reduced gas crossover (hydrogen permeability below 1.25 × 10−4 cm3/s/cm2 in Nafion membranes), and wide operating temperature range (20–80 °C), making them highly efficient for hydrogen production.19–21 A critical component of PEMWEs is the catalyst used for oxygen evolution reactions (OERs), where iridium oxide (IrO2) is widely regarded as the state-of-the-art catalyst due to its excellent stability and activity. In contrast, ruthenium oxide (RuO2) demonstrates a higher initial catalytic activity but suffers from rapid degradation, losing up to 40–50% of its activity within 10–20 h of OER operation, whereas IrO2 maintains over 90% of its activity even after 100 h, making RuO2 less practical for long-term use.22–25 On the cathode side, platinum is employed as the benchmark catalyst due to the efficiency for hydrogen evolution reactions (HERs), and its optimal hydrogen adsorption-free energy (ΔGH* ≈ 0), which facilitates efficient hydrogen adsorption/desorption leading to low overpotentials (<30 mV vs. reversible hydrogen electrode (RHE)).26–28
On the other hand, PEMFCs are recognized for their clean energy conversion capabilities, characterized by high power density (0.5–1.0 W/cm2), energy efficiency (∼40–60%), and rapid start-up times while operating at low temperatures (60–90 °C).29–32 Platinum is used as the primary catalyst in PEMFCs due to its exceptional stability and high catalytic activity, with specific activities exceeding 0.3 A/mg Pt for oxygen reduction reactions (ORRs) and typical loadings around 0.1 mg Pt/cm2, facilitating efficient hydrogen oxidation reaction (HOR) at the anode and oxygen reduction at the cathode.33,34 Despite their advantages, PEM technologies experience significant challenges, particularly related to the high cost and degradation of electrocatalysts, which remain major obstacles to their widespread commercialization.35,36 Over time, the degradation of these catalysts leads to spent components that contain valuable but underutilized precious metals. Consequently, there is a growing need for the development of efficient recovery methods to reclaim critical metals from spent PEM technologies, which are both cost-effective and environmentally friendly.37,38
In PEM technology, dissolution of electrocatalysts is an undesirable process, as it leads to loss of active surface area, degradation of electrode structure, and decreased performance of the system. Despite significant advancements in catalyst design, ensuring long-term stability under harsh electrochemical conditions remains a challenge. 39 This is particularly important for platinum and iridium, which are scarce and expensive electrocatalysts, where even small losses increase material replacement costs and hinder large-scale application. 40 As a result, researchers have developed methods to recover electrocatalysts from spent PEM materials, often using controlled conditions that induce electrocatalyst dissolution similar to those occurring in PEM technologies. This review summarizes the dissolution of Pt and IrO2 electrocatalysts from spent PEM technologies using both conventional and emerging recovery methods. Conventional techniques, such as hydrometallurgy, have been widely employed for metal recovery but it often involve harsh chemical treatments and complex separation processes. 41 In contrast, an electrochemical dissolution method presents a promising alternative, offering greater selectivity, reduced environmental impact, and potential scalability. The effects of parameters such as potential range, scan rate, electrolyte composition, and pH are explored, as they play a significant role in influencing dissolution efficiency and metal recovery rates.
Pt and Ir in PEMFC and PEMWE systems
PEM technology plays a pivotal role in modern electrochemical energy systems; Figure 1 illustrates the fundamental working principles of a PEMFC and a PEMWE. In a PEMFC system (Figure 1(a)), chemical energy stored in hydrogen is converted into electrical energy through electrochemical reactions.42–46 At the anode, the HOR occurs, where hydrogen molecules split into protons and electrons. At the cathode, the ORR occurs, where oxygen combines with protons and electrons to produce water and heat. 47 In contrast, a PEMWE system (Figure 1(b)) uses electrical energy to split water into hydrogen and oxygen gases.48–50 At the anode, water is oxidized to produce protons, electrons, and oxygen gas. The formed protons then migrate through the membrane to the cathode, where they are reduced, leading to the production of hydrogen gas (H2).42,51
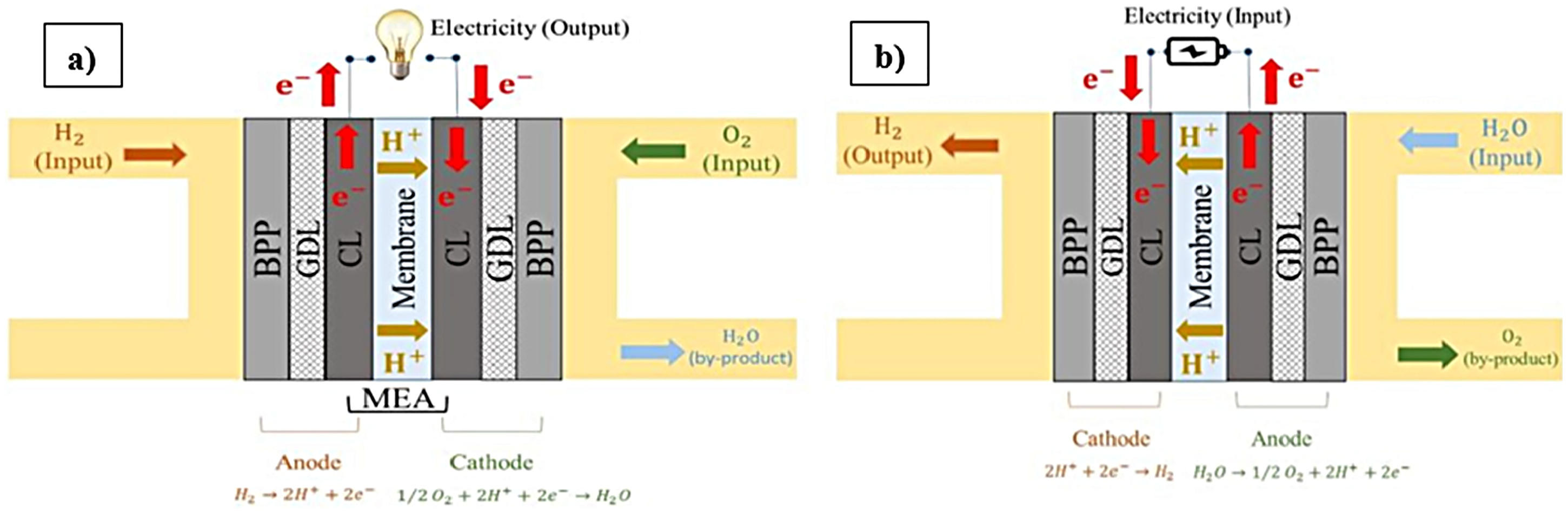
Schematic of a PEMFC (a) and PEMWE system (b) with all the required layers in a cell. 42 (Reproduced with permission from A. Baroutaji et al., under Copyright Clearance Center, Inc. (CCC), International Journal of Hydrogen Energy). PEMFC: proton exchange membrane fuel cell; PEMWE: proton exchange membrane water electrolyzer.
Equations (1), (2), and (3) represent the key electrochemical reactions in PEMFCs, including the HOR at the anode, the ORR at the cathode, and the overall cell reaction:

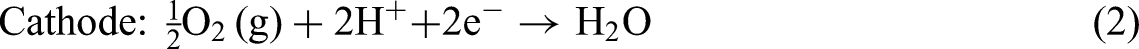
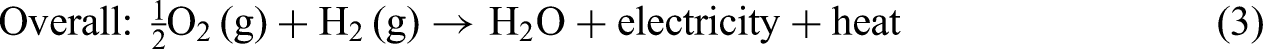
In PEMWEs, the efficiency of the water-splitting process is highly dependent on the chosen electrocatalysts, with platinum serving as the state-of-the-art catalysts at the cathode and ruthenium oxide (RuO2) or iridium oxide (IrO2) at the anode.
6
These catalysts lower the activation energy required for HERs and OERs, with their performance evaluated based on activity, stability, and efficiency.
52
Equations (4), (5), and (6) summarizes the fundamental electrochemical reactions in PEMWEs, including the OER at the anode, the HER at the cathode, and the overall water-splitting reaction:
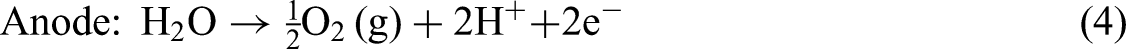

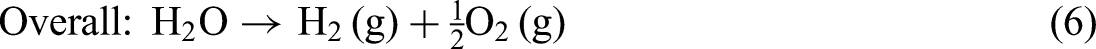
The key component of PEM technology is a membrane electrode assembly (MEA), which consists of a proton-conducting membrane that is sandwiched between catalyst layers.43,53 MEA serves multiple functions, which include acting as a barrier to separate reactants, an electrical insulator to direct current through an external circuit, and a proton conductor to facilitate the movement of protons between the anode and cathode. 7 Catalyst layers form the core of the MEA, as they are the sites where electrochemical reactions take place.54–56 The noble electrocatalysts particularly platinum and iridium, though essential for high catalytic activity, are highly susceptible to degradation when exposed to high temperatures, acidic environments, and high potentials during operation, leading to the failure of the MEA.57–59 In addition to catalyst degradation, other mechanisms such as carbon support corrosion and electrocatalyst particle sintering further accelerate the loss of active surface area and structural integrity of the catalyst layer, leading to MEA failure, ultimately reducing the operational lifetime of PEM technology.60–62
Despite their critical role, platinum group metal (PGM) electrocatalysts are not immune to degradation under operational conditions. Prolonged exposure to elevated temperatures can lead to the accelerated dissolution of Pt and Ir from the catalyst surface, decreasing the number of active sites available for the desired electrochemical reactions, such as ORR and HER. 48 Similarly, cycling at high potentials, especially under conditions typical in fuel cell or electrolyzer operations, can lead to metal dissolution, particle agglomeration, and sintering, further compromising catalyst performance.49,50 Chloride impurities, introduced either through airborne contamination, water supply, or residuals in catalyst and membrane materials, have also been identified as critical contributors to catalyst degradation in PEM systems.63,64 In addition to catalyst degradation, the ion conductivity of the membrane itself can deteriorate over time due to mechanical stress, chemical attack, and water management issues, which further reduces the efficiency of the MEA. 52 These degradation mechanisms contribute to the overall performance decline of PEM systems, often necessitating expensive replacements and extensive maintenance. Understanding and mitigating these limitations through improvements in catalyst stability, membrane durability, and system design is essential for increasing the operational lifetime and cost-effectiveness of PEM technologies.65,66
PEMFC and PEMWE electrocatalysts
Platinum is widely used as the benchmark catalyst in PEMFCs for redox reactions due to its exceptional catalytic efficiency and stability, with a Pt/C specific activity of ∼200 µA/cm2 for ORR and electrochemical surface area (ECSA) loss less than 40% according to the Department of Energy durability target.38,67,68 It exhibits optimal binding energy and high electrocatalytic mass activity (0.1–0.3 A/mg Pt at 0.9 V).68,69 However, apart from the high cost and limited availability, research shows that platinum electrocatalysts can redeposit in PEM systems leading to performance degradation and reduced efficiency of PEM systems over time.70–72 Additionally, platinum nanoparticles have been observed to diffuse into the cathode catalyst layer and accumulate in the membrane, reducing the effectiveness of the nanoparticles as a catalyst.70,73 The combination of these factors has significantly reduced the operational lifetime of PEMFCs. Currently, due to the failure of the MEA, PEMFCs have an estimated lifespan of 10 years.74,75
In PEMWEs at the anode, iridium oxide is the preferred electrocatalyst over ruthenium oxide due to its superior stability in acidic environments, along with remarkable durability and activity, which are essential for OERs.65,66,76,77 However, the anodic catalyst layer typically uses 2–4 mg/cm2 of iridium, while the cathodic catalyst layer only requires 0.2–0.4 mg/cm2 of Pt/C, contributing to the high cost of the technology. 78 While PEMWEs are highly effective at producing high-purity hydrogen, several challenges limit their broader commercialization. The primary limiting factors include membrane poisoning and performance loss caused by the dissolution of the anode and cathode electrocatalysts at high potentials.66,76,79 Overall, both platinum and iridium oxide can experience increased dissolution rates under conditions such as low catalyst loadings, high current densities, and high operating potentials, which poses a challenge for maintaining long-term electrode stability and limits the broader commercialization of PEMWEs.77,80 PEMWEs exhibit degradation pathways similar to PEMFCs, including catalyst dissolution and detachment. Under certain conditions, degradation can also proceed during open-circuit voltage periods, particularly for platinum-based catalysts.75,79 While platinum may undergo limited chemical dissolution when exposed to acidic electrolytes under open-circuit conditions, iridium dissolution under such conditions is generally negligible.78,81,82
To address the issue of unusable catalysts that accumulate postoperation, efforts of research and development have been directed toward the development of innovative solutions to recycle these electrocatalysts from spent PEM technologies.83–87 Such strategies include chemical leaching, electrochemical dissolution, and physical separation methods, all aimed at efficiently extracting platinum, iridium, and other precious metals without compromising their activity for subsequent reuse.88–91 These efforts not only ensure a sustainable supply of precious metals but also reduce the environmental and economic impacts of traditional mining and refining processes.92–94 Furthermore, directly reusing recovered electrocatalysts from spent hydrogen fuel cells provides a highly sustainable and cost-effective alternative, promoting circular economy models and encouraging innovation in materials design, process optimization, and renewable energy conversion technologies while minimizing the overall carbon footprint of the energy sector.95,96
Platinum and iridium dissolution in hydrogen technologies
The selection of electrocatalysts for electrochemical processes is crucial in renewable energy technologies such as PEMWEs and PEMFCs, with the ideal electrocatalyst required to possess good stability and high catalytic activity.97–99 Despite their favorable catalytic properties, Pt and Ir electrocatalysts face significant challenges with dissolution during operation, which affects their performance and hinders the widespread application in renewable energy technologies. Yu et al. analyzed the distribution of Pt and Ir in a degraded PEMWE MEA after 4500 h operation, where before testing, the catalyst-coated membrane had an active geometric area of 86 cm2. The anodic catalyst layer consisted of iridium oxide (IrO x ) nanoparticles with a loading of 0.08 mg/cm2, while the cathodic catalyst layer consisted of Pt nanoparticles supported on Vulcan® XC-72R carbon black with a Pt loading of 0.3 mg/cm2. 77 During electrolysis testing, significant iridium dissolution occurred at the anode, where only 30% of the initial IrO x remained in the anodic catalyst layer. The IrO x migrated through the membrane, forming an oxide band near the anode and depositing in the membrane (18%) and at the cathode (42%), where it was reduced by crossover hydrogen or coprecipitated with Pt. Platinum dissolution was observed to be less severe, with about 10% migrating primarily into the membrane, where it forms Pt–Ir particles mostly near the cathode side as illustrated in Figure 2.77,100
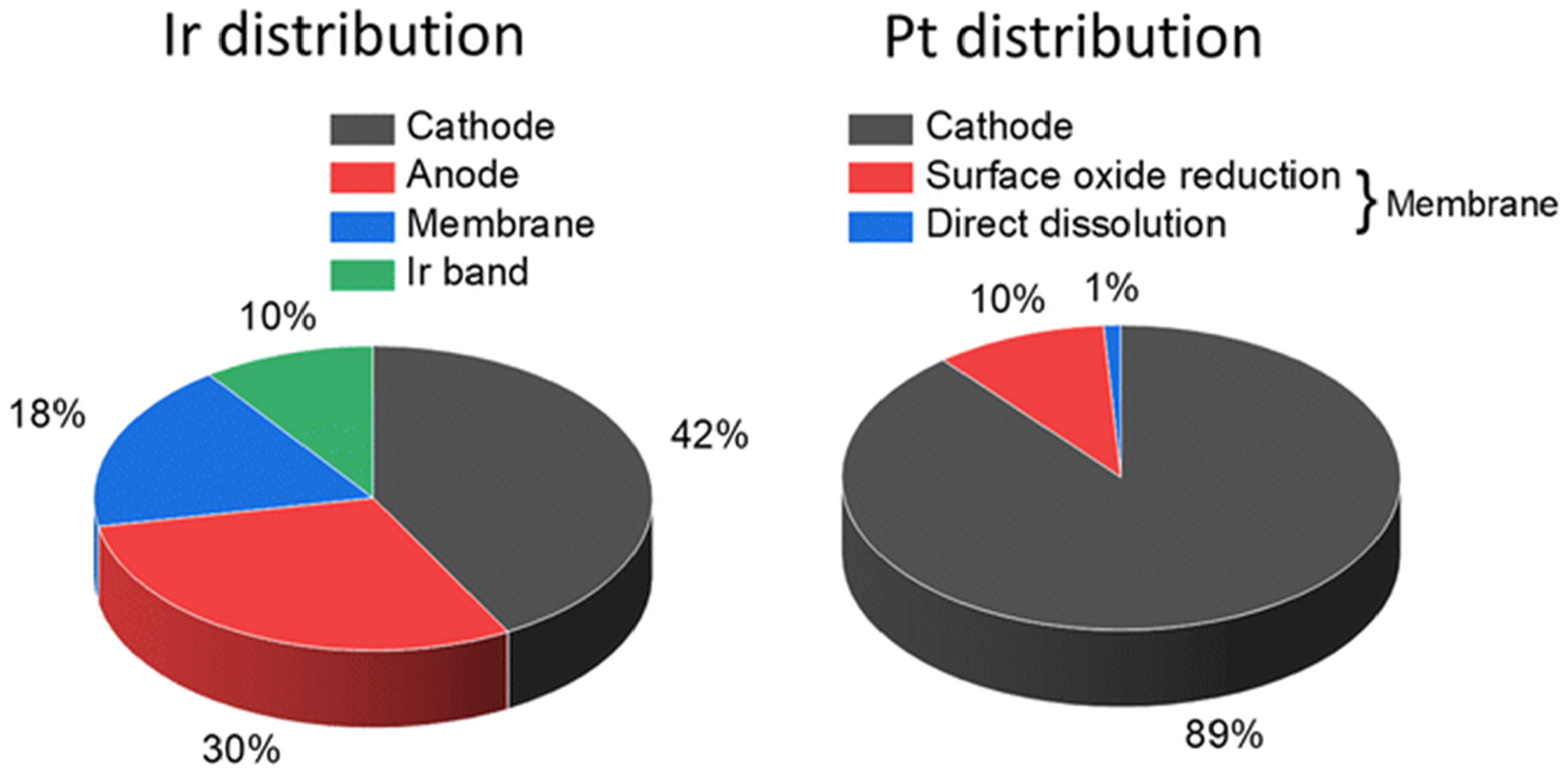
Distribution of iridium and platinum postmortem investigation of a PEMWE MEA with a low catalyst loading of 0.3 mg/cm2 Pt and 0.08 mg/cm2 Ir after a long-term test of 4500 h at 1.8 A/cm2. 100 (Reproduced with permission from Hans Becker et al., under Creative Common (CC BY) license (https://creativecommons.org/licenses/by/3.0/)). PEMWE: proton exchange membrane water electrolyzer; MEA: membrane electrode assembly.
Platinum dissolution mechanism
Electrochemical dissolution of platinum occurs through a transient mechanism, driven by the interaction between the oxidation and reduction processes, leading to the formation and reduction of Pt oxides. 101 One of the widely proposed dissolution pathways is the place exchange (PE) mechanism, in which the Pt surface undergoes alteration due to the electrochemical formation and reduction of an oxide layer. 102 In the PE mechanism, a Pt atom is displaced from the top surface layer, while maintaining its in-plane lattice position. The resulting vacant site is then occupied by an oxygen atom adsorbed on the surface previously, as illustrated in Figure 3.103–105 Under mild electrochemical conditions, platinum is oxidized to form a Pt oxide (oxide I) monolayer. However, under more severe electrolytic conditions, a multilayer Pt oxide (oxide II) is formed.106,107 PtO can diffuse across the Pt surface to energetically favorable sites, increasing the exposure of underlying Pt atoms to the electrolyte. This exposure of Pt atoms to the surface increases the likelihood of dissolution, as Pt exhibits high surface energy, making it more prone to dissolution.108–110
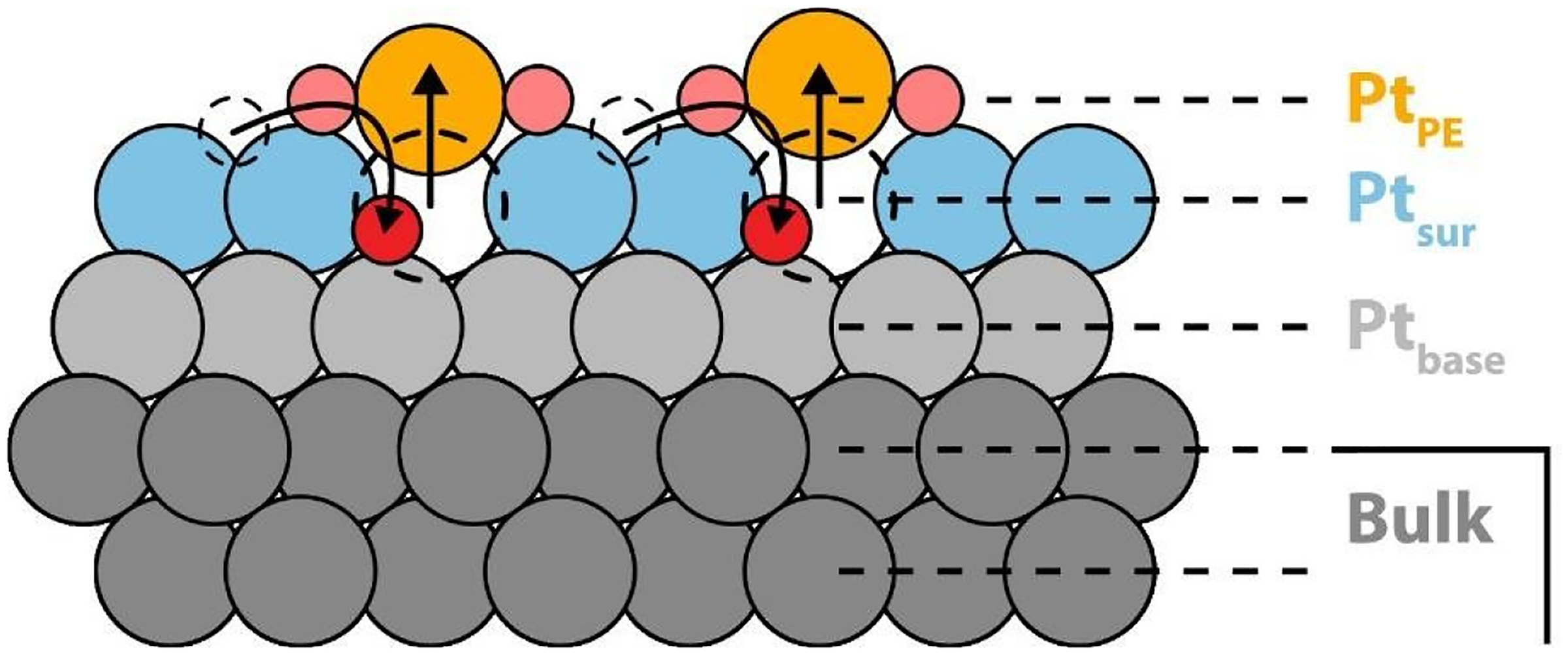
Oxide place exchange model of Pt under mild conditions. (Reproduced with permission from authors under Creative Common (CC BY) license (https://creativecommons.org/licenses/by-nc-nd/4.0/)). 103
Iridium dissolution mechanism
Iridium dissolution is initiated by changes in oxidation state, particularly during the initial reduction of the anhydrous IrO x oxide layer. The participation of iridium oxides in the OER has been shown to promote dissolution, with higher OER activity accelerating the rate of iridium loss.111–113 During OER on iridium catalysts, several dissolution mechanisms are possible, including direct dissolution of metallic Ir, dissolution through Ir(V)–Ir(III) transition, and formation of IrO3 at high potentials. The dissolution route through IrV–IrIII transformation dominates for metallic iridium.114–116 Figure 4 illustrates the universal scheme for OER and Ir dissolution, where the OER and dissolution mechanism are intertwined through a common reaction intermediate. In the scheme, the green arrows indicate the most favorable mechanism for iridium-based materials, where OER occurs at lower potentials. The red arrows indicate the dominant dissolution route at higher anodic potentials, while the blue arrows represent intermediate steps that occur regardless of the electrode material and potential. 116
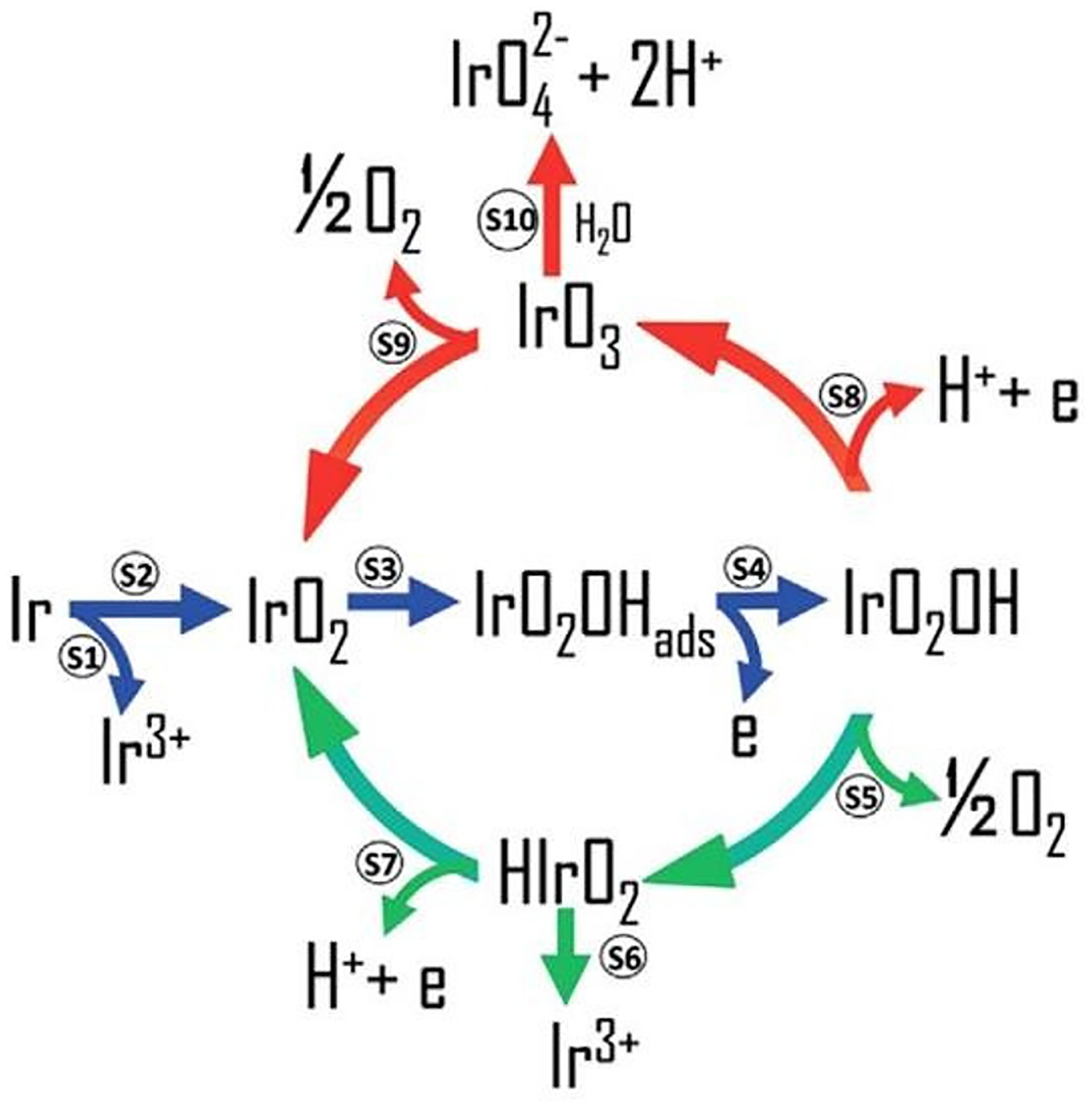
Universal mechanism correlating both the OER and dissolution pathways. 116 (Reproduced with permission from Olga Kasian et al., under Creative Common (CC BY) license (https://creativecommons.org/licenses/by-nc-nd/4.0/)). OER: oxygen evolution reaction.
PGM recovery methods
Conventional methods
Due to the cost of PGMs, researchers have been actively exploring methods to recover these valuable metals from MEAs. However, recovering PGMs from PEM systems presents significant challenges due to the required specialized recycling processes. The recycling of the membrane and PGMs typically involves shedding the used MEA and delaminating its various layers. 6 Currently, the primary end-of-life (EoL) technologies for recovering platinum and iridium from composite materials are hydrometallurgy and pyrometallurgy.7,117 Table 1 summarizes the conventional recovery technologies used to recover PGMs from PEM technology.
Conventional recovery methods for critical materials in PEMFC and PEMWE systems.

PEMFC: proton exchange membrane fuel cell; PEMWE: proton exchange membrane water electrolyzer.
Pyrometallurgical processes are the primary method to recover PGMs from spent PEM technologies, typically consisting of three main processes: sintering, chlorination, and smelting. 5 However, the vaporization process that occurs during the chlorination and smelting processes contributes to atmospheric pollution. Additionally, pyrometallurgy poses health and environmental risks due to using hazardous gases such as Cl2 and CO.5,118 In contrast, hydrometallurgy has emerged as the leading recovery process, as it is regarded as a more favorable alternative to pyrometallurgy.6,119 The hydrometallurgy process consists of two main steps: (i) the leaching step, where a mixture of strong acid and oxidizing agent is used to dissolve metals from ores in an aqueous solution, and (ii) the separation step, where solid carbon particles are filtered out from dissolved PGMs.6,120–122 While hydrometallurgy is frequently described as an environmentally friendly process compared to pyrometallurgy, it still has significant drawbacks. The process generates harmful effluents that contribute to water pollution, and solid waste that negatively impacts the environment. 119 Additionally, toxic byproducts such as NO x gases remain a major concern associated with associated with hydrometallurgy (Figure 5). 123
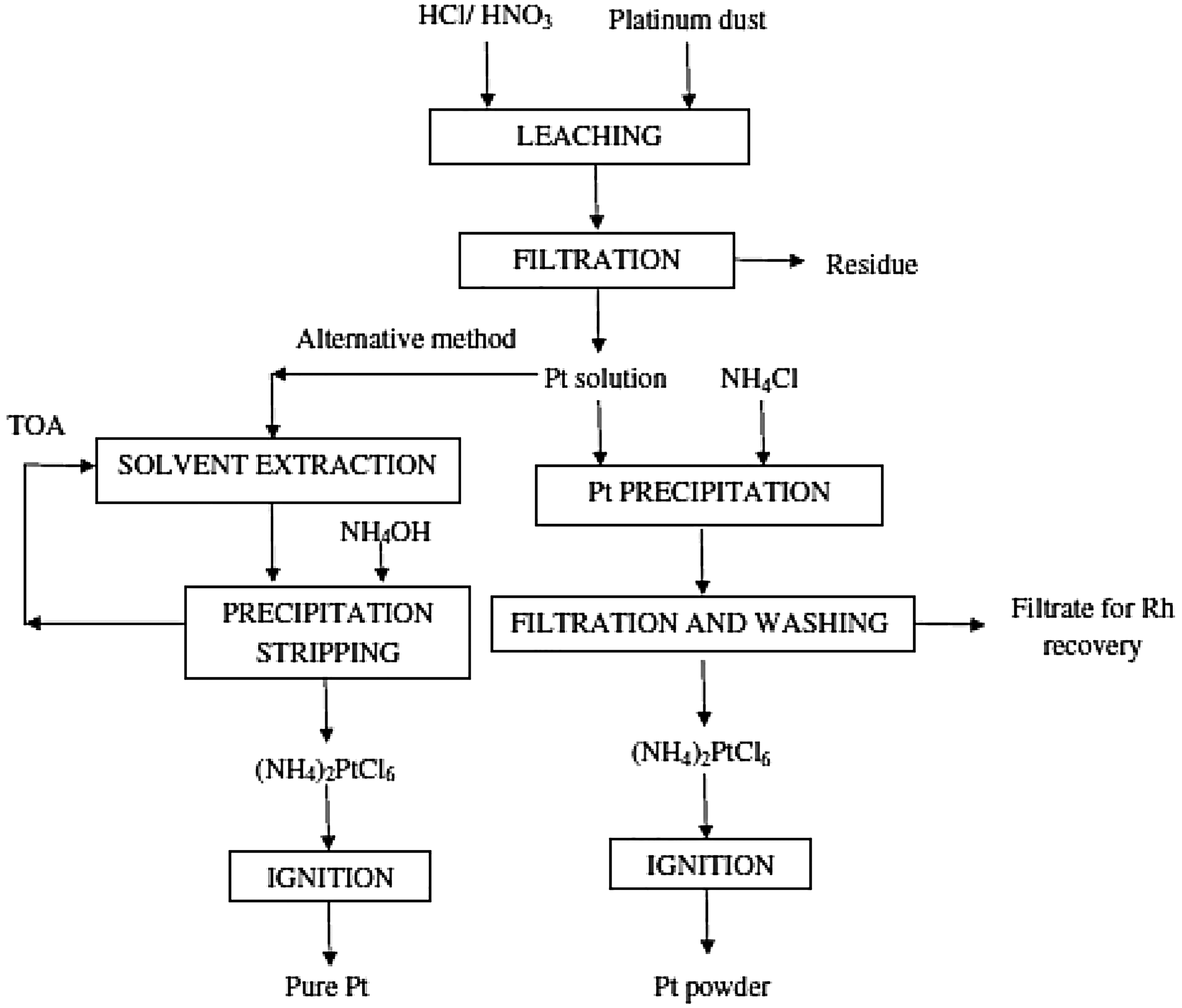
Flow chart for the recovery of platinum by hydrometallurgical process from platinum dust. 119 (Reproduced with permission from Elsevier, license number: 5491221407891).
Electrochemical dissolution methods
PGMs such as platinum and iridium are valued for their exceptional catalytic properties, making them critical in various industries, particularly in renewable energy technologies. 85 However, at the end of the lifecycle of hydrogen fuel cells, PGMs remain embedded in the spent MEAs and are often rendered waste. 7 The growing accumulation of spent electrocatalysts has prompted an urgent need for efficient and sustainable recovery strategies to minimize environmental impact. Traditional recovery methods, such as pyrometallurgical and hydrometallurgical processes, often require extreme temperatures, aggressive chemicals, and produce significant waste. 124 As a result, attention has increasingly turned toward innovative, low-impact approaches that align with circular economy principles. In this context, selective electrochemical dissolution has emerged as a promising recovery method, effectively dissolving PGMs such as platinum and iridium into their ionic forms while consuming less energy and fewer harmful chemicals. 84
Several strategies have been explored to recover platinum and iridium from spent MEAs through electrochemical dissolution, followed by reduction of the dissolved ionic species into their metallic or precursor forms, thereby closing the materials loop for catalyst reuse.87,92 For instance, Sharma et al. demonstrated the electrochemical dissolution of platinum from spent MEAs in dilute hydrochloric acid, leading to the formation of soluble platinum chlorocomplexes such as (NH4)2PtCl6, which were subsequently reduced in an ethylene glycol and water mixture to form Pt/C nanoparticles. The recovered Pt/C exhibited ECSA and activity comparable to commercial catalysts when evaluated under accelerated stress testing. 125 In a related study, Sharma et al. also reported an environmentally friendly electrochemical recycling process in which platinum nanoparticles were dissolved through potentiodynamic cycling in mildly acidic electrolytes and subsequently recovered using electrodeposition onto another substrate within the same electrochemical system, achieving efficient Pt recovery. 126 Furthermore, Sakthivel et al. introduced a one-pot cathodic leaching–redeposition system in 2 M HCl, where electrochemical cathodic leaching and electrochemical cathodic deposition occurred sequentially in the same cell. This approach achieved nearly complete Pt recovery from spent gas diffusion electrodes, and the redeposited Pt was confirmed by SEM and TEM analyses to consist of uniform metallic nanoparticles with high purity. 127
Recent studies have explored various methods for the dissolution and recovery of iridium, a precious metal widely used in electrochemical applications. Moriau et al. investigated the electrochemical dissolution of iridium in acidic media, demonstrating a significant increase in dissolution rates when chloride ions and organic molecules, such as ethanol, were present. 114 Sharma et al. further examined the electrochemical dissolution of iridium oxide (IrO x ) in the presence of metal ions like Cu2+, revealing an enhancement in dissolution rates by approximately threefold. 128 Additionally, an electrochemical dissolution method of high-purity iridium powder recovery was developed, directly in concentrated hydrochloric acid to produce a chloro-iridic acid (H3IrCl6) solution. The method utilizes a U-shaped acid-resistant electrolyzer charged with iridium powder and 8–12 M HCl at 100–115 °C for 1–15 h, with the resulting solution yielding high Ir (≥99.9%). 129 Collectively, these studies highlight diverse strategies for iridium dissolution using electrochemical methods; however, they primarily focus on quantifying dissolved Ir and characterizing residual materials, and not on the actual recovery.
Potential cycling and square wave
Studies have demonstrated that all PGMs undergo transient dissolution when subjected to potential cycling within a window that allows oxidation and oxide reduction. Platinum and iridium dissolution are strongly potential-dependent processes, with dissolution rates accelerating at higher anodic limits. Platinum dissolution primarily occurs at potentials above 1.1 V RHE where the formation and subsequent reduction of oxides destabilize surface atoms, leading to transient dissolution.91,112 At these potentials the cathodic scan becomes the dominant phase in platinum dissolution, due to the PtO2 formed at high potentials being unstable during reduction.130,131 For iridium, dissolution becomes significant when cycling up to an anodic limit of 1.385 V RHE under oxygen evolution conditions. At these potentials, iridium atoms at the surface of the catalyst repeatedly cycle between multiple oxidation states. These continuous redox transitions destabilize the iridium oxide lattice, weakening the surface and ultimately causing iridium atoms to detach and dissolve into the electrolyte as soluble species. Here, the anodic scan is the dominant phase in dissolution.112,132,133
Multiple studies on potential cycling protocols provide direct evidence for dissolution of Pt and Ir under redox cycling. For instance, Yasuda et al. studied Pt/C MEA catalysts under anode/cathode potential cycling and found that Pt dissolution and redeposition in the membrane layer were triggered by cycling into the Pt oxide formation and reduction potential regions. 134 Ali-Löytty et al. demonstrated on Pt(111) that transient dissolution occurs during the reduction phase of the oxide formed at high potentials. 135 In fuel-cell start/stop cycling of Pt catalysts, Ettingshausen et al. found that cycling through oxidation/reduction steps caused markedly higher Pt loss than static holds. 136 Sugawara et al. quantified that Pt dissolution under cycling is strongly increased when the upper potential limit (UPL) is above ∼0.8 V RHE and the lower limit allows full oxide reduction (below ∼0.6 V RHE). 137 On the other hand, Jovanovic et al. applied potential cycling from ∼0.05 to varying upper potentials (up to ∼1.6 V vs. RHE) and observed that Ir dissolution increases with the UPL, consistent with oxide redox-driven lattice destabilization. 138 More recently, Geuß et al. quantified Ir dissolution under realistic OER potential cycling conditions, highlighting that repeated oxidation and reduction cycles critically influence the oxide state and promote the detachment of Ir atoms. 81 A recent review by Becker et al. highlights that Ir dissolution correlates with both the applied potential window and the thickness/structure of the oxide layer, with an increase in both parameters leading to enhanced dissolution. 100
The dissolution of PGMs is significantly influenced by the applied waveform, with pulse-based techniques such as square wave voltammetry (SWV) being considered a more efficient technique for PGM dissolution due to its faster processing time, peak-shaped response, and enhanced sensitivity. The main principle behind its enhanced sensitivity is due to the difference in the decay rates of the faradaic and the nonfaradaic current.118,139–142 In SWV a combination of a staircase and a square potential waveform is used, thereby repeatedly driving the electrode surface into oxide-formation and subsequent oxide-reduction regimes, promoting transient dissolution of PGMs as indicated in Figure 6(b). In contrast to the gradual potential sweep used in conventional cyclic voltammetry, SWV applies sharp and frequent potential changes. These rapid transitions generate transient structural changes causing the release of metal ions into the electrolyte.91,130
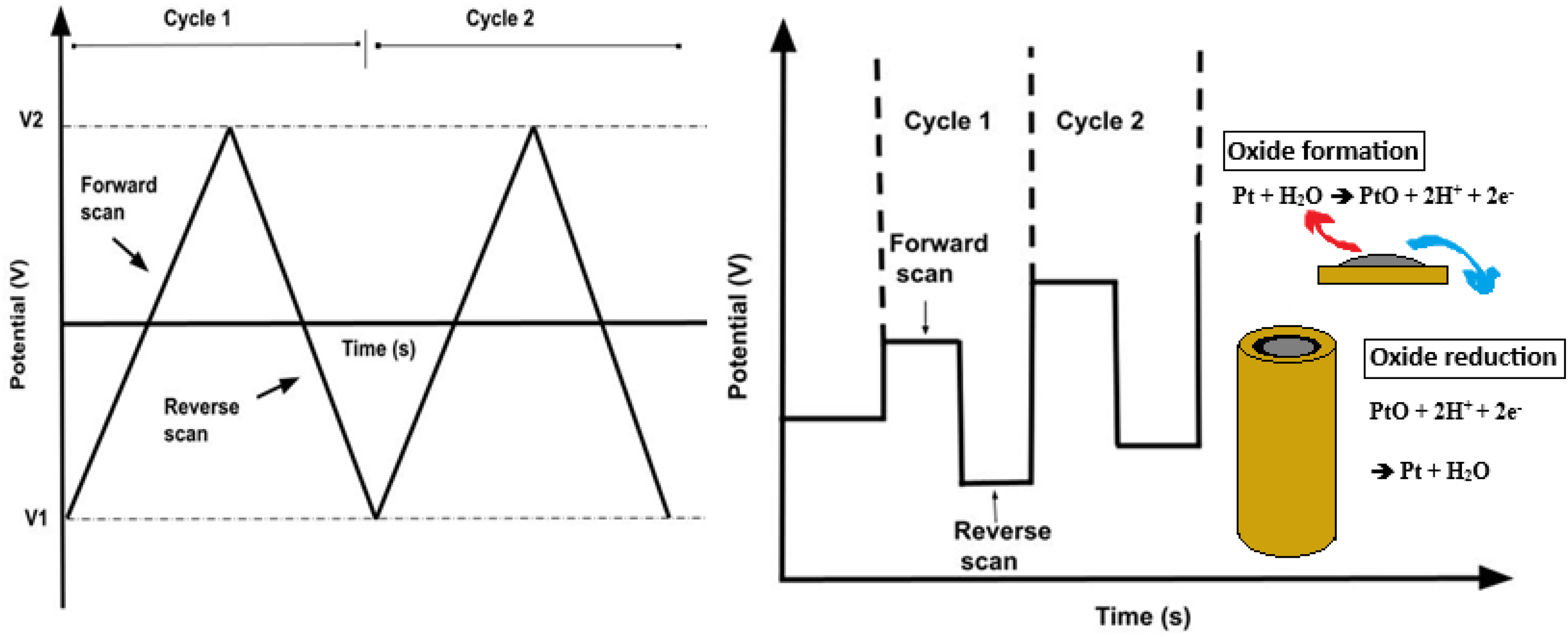
Electrochemical dissolution techniques: cyclic voltammetry (a) and square wave voltammetry (b).
Although only a limited number of studies have examined PGM dissolution using SWV, the available findings demonstrate its significant potential for enhancing dissolution efficiency. Kanamura and Yagyu developed a method that recovered 93.2% of Pt from an MEA catalyst loading of 0.91 mg/cm2, using SWV in 1 M HCl electrolyte, with alternating reduction and oxidation potentials of 0.1 and 1.5 V versus RHE. 143 Similarly, Percival et al. achieved a Pt dissolution rate of 0.173 ng/(cm2/s), and a total dissolution of 26.25 ng/cm2, after 150 square wave pulses in 100 mM KCl, applied potentials between 0 and −1 V versus Ag/AgCl. 144 In the case of iridium, dissolution has been observed during the shut-up/shut-down half-cycles, where the anode potential oscillates in a square-wave form between 0 and 1.05 V versus RHE potential, leading to the formation of soluble Ir n + species. 140 These findings demonstrate that SWV can be precisely tuned to induce accelerated dissolution of Pt and Ir. While such dissolution is undesirable during normal PEM technology operation, it becomes advantageous when repurposed as a recovery strategy.
Effect of electrolyte composition and pH on dissolution of Pt and Ir
PGM electrocatalysts are generally perceived as stable and inert, essential for facilitating chemical reactions without undergoing significant changes. However, these noble metals have been observed to leach in harsh alkaline and acidic environments due to various effects attributed to the anions and cations present in the electrolyte. The dissolution of platinum and iridium is strongly influenced by the nature and concentration of the electrolyte, affecting the formation of soluble metal species in the electrolyte. 145 Dissolution rates under acidic conditions have been reported to be higher than that in alkaline media, due to proton (H+) assisted destabilization of surface oxides, whereas in alkaline electrolytes, the higher concentration of hydroxide ions (OH−) leads to the formation of more stable, protective metal hydroxide layers that stabilize the surface and inhibit further dissolution.146,147
Chloride-containing electrolytes have been employed in PGM dissolution studies because they not only promote dissolution but also act as complexing agents. In these electrolytes, Pt and Ir dissolution are chemically assisted, where oxidized Pt and Ir ions bind to chloride ions, forming stable soluble complexes such as [PtCl4]2−, [PtCl6]2− and [IrCl6]3−, [IrCl6]2−. These complexes prevent redeposition and thereby promoting dissolution.64,107,114,148 At potentials below 1.0 V, Cl− enhances Pt dissolution primarily through the formation of the [PtCl4]2 complex with the rate of dissolution increasing with higher Cl− concentration. At potentials above 1.2 V, dissolution becomes dominated by the formation of the [PtCl6]2− complex where the surface of passivating Pt–O layer is chemically attacked by Cl− removing the oxide layer, allowing continuous formation of [PtCl6]2− and increasing dissolution.141,149 Figure 7 illustrates a multistep mechanism of how Pt dissolves in a chloride-containing electrolyte. Initially, a surface Pt atom undergoes oxidation to form a Pt–O species, which destabilizes its bonding with the underlying Pt lattice. While the protective oxide layer shields the underlying metal from further oxidation, the chloride ions interact with the oxidized Pt, hindering the stability of this oxide layer. The oxidized Pt atom detaches from the surface and dissolves into the electrolyte as Pt4+ ions, stabilized by chloride ligands such as [PtCl6]2− causing Pt dissolution.64,107,141
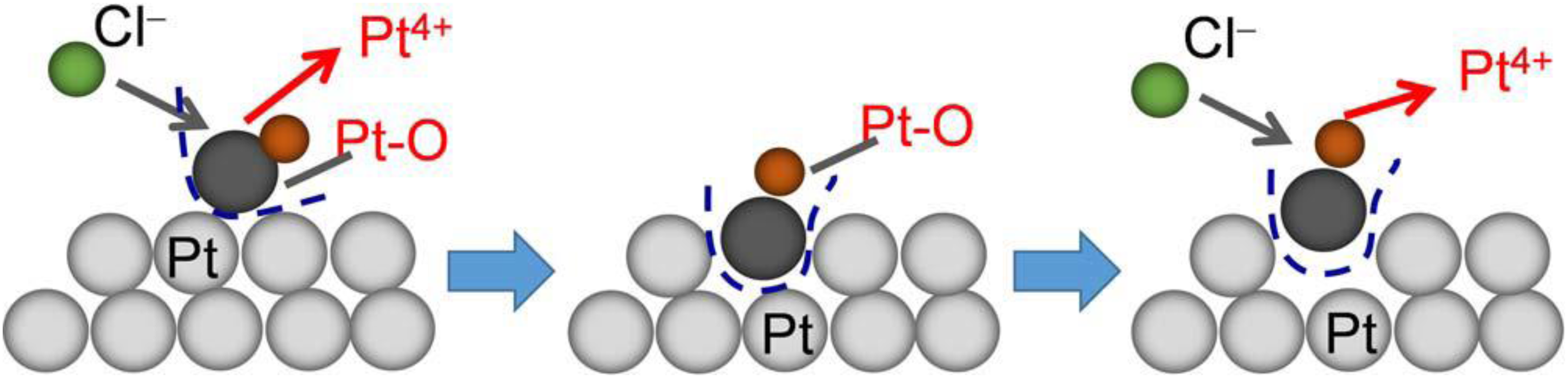
Dissolution mechanism of Pt4+ in the presence of Cl− under potential cycling conditions. 141 (License ID: 1644908-2).
Quantitatively, Latsuzbaia and coworkers reported 22.5 µg/cm2 of Pt per cycle in 0.1 M HCl for cycling between 0.5 and 1.1 V versus RHE at 50 mV/s under controlled mass transfer, achieving full nanoparticle dissolution within 3–5 h for ∼0.35 mg/cm2 loadings.
83
In a more concentrated chloride bath, Sharma et al. measured ∼30 µg of Pt per cycle in 1 M HCl for 0.4–1.6 V versus RHE at 100 mV/s on PEMFC electrodes, with the dissolved Pt recovered as (NH4)2PtCl6 and later chemically reduced to synthesize Pt/C electrocatalysts.
150
Mechanistically, in-situ channel-flow studies showed that Cl− accelerated anodic dissolution above 1.2 V versus standard hydrogen electrode (SHE) by enhancing [PtCl6]2−/[PtCl4]2− formation and increased cathodic dissolution during PtO2 reduction.
141
Using a channel-flow multi-electrode system Wang et al. reported the effect of Cl− on Pt dissolution under both potentiostatic and potential cycling. The presence of chloride ions in 0.5 M H2SO4 significantly enhanced the dissolution of Pt under both potentiostatic and cycling conditions. At potentials below 1.0 V versus SHE, the effect was modest, whereas at potentials above 1.2 V versus SHE, dissolution increased by more than an order of magnitude relative to chloride-free conditions. The study reports the formation of
These studies establish chloride as a key promoter of noble-metal dissolution by stabilizing oxidized metal–chloro complexes and suppressing oxide passivation. This mechanistic role of chloride extends beyond Pt and is also observed for Ir-based catalysts. The strong stability of the IrO2 passive layer under normal operational conditions limits iridium dissolution. However, during potential cycling in chloride-containing electrolytes, iridium dissolution becomes thermodynamically favorable due to the formation of soluble chloro-complexes such as [IrCl6]3− and [IrCl6]2−, which effectively stabilize oxidized Ir3+/Ir4+ species in solution.114,152,153 At potentials above approximately 1.0 V versus RHE, the dissolution rate accelerates due chloride ions adsorbing onto the IrO2 surface and penetrating the oxide lattice, weakening Ir–O bonds through ligand exchange and forming Ir–Cl intermediates.138,154,155 This process disrupts the integrity of the passive oxide layer as it facilitates the formation of Ir x Cl y soluble complexes. The dissolution pathway is further enhanced under repetitive redox cycling, as the continuous oxidation and reduction of Ir species creates transient surface states that favor chloride coordination and solubilization.81,112,114
The absence of complexing anions such as Cl− results in extremely low dissolution, making recovery via precipitation or electrodeposition inefficient and often below analytical detection limits. 114 The effect of chlorides was studied by Moriau et al. using a H-cell electrochemical system in the potential range 0–1.385 V versus RHE, it was shown that the presence of chlorides benefits the leaching of iridium, with a 10-fold increase for HCl over HClO4 and a 40 times enhancement in 1 M ethanol and HCl solution compared to a HClO4 electrolyte. However, the overall iridium dissolution was low at 0.91% in 0.1 M HCl and 0.08% in 0.1 M HClO4 after 100 cycles. 114 In a more concentrated 1 M HCl electrolyte, Sharma et al. studied the effect of transition metal ions on IrO x dissolution. Dissolution was enhanced by threefold in the presence Cu2+ metal ions during 0.0–1.65 V cycling at 100 mV/s. Structural characterization performed on the samples collected from glassy carbon rotating disk electrode showed Ir dissolution up to 40% where the elemental distribution of Ir–O remained relatively uniform after cycling. 128
More recently, studies have evaluated dissolution of both platinum and iridium under nonchloride conditions, which primarily proceeds through oxide-mediated mechanisms rather than halide complexation.107,114,156 Pt dissolution in chloride-free electrolytes such as perchloric and sulfuric is largely driven by the formation and reduction of its oxide layer via a place-exchange mechanism illustrated in Figure 3, where surface Pt atoms are destabilized during repeated formation and reduction of surface oxides, releasing Pt2+/Pt4+ into solution.75,80,91 For iridium, dissolution in Cl−-free electrolytes such as HClO4 leads to gradual oxidative detachment of surface Ir atoms during the Ir3+/Ir4+ redox transitions, with dissolution strongly correlated with the formation and reduction of transient IrO x species. This process is well described by the universal mechanism, which correlates OER pathways with the coupled dissolution of active Ir sites through transient oxide formation and reduction.114,138 Studies conducted in nonchloride electrolytes have offered insight into the oxide driven dissolution mechanism of Pt and Ir; however, these studies are limited by the inherently low solubility of the dissolved species, which restricts quantitative recovery of Ir and Pt.91,157,158
Several studies have explored intrinsic factors affecting dissolution in these nonchloride acidic environments, highlighting the key roles of pH and oxide-mediated mechanisms. Flow-cell studies by Topalov et al. demonstrated that Pt dissolution onset (∼1.1 V vs. RHE) is largely independent of anion identity, but cathodic dissolution during oxide reduction is strongly influenced by proton concentration. 91 Yadav et al. further confirmed that dissolved Pt in buffered acidic solutions is minimal at moderate pH, where the amount of dissolved Pt was almost negligible in a pH 3.0 solution, while the ECSA loss in this solution was nearly identical to that observed at pH 0.4, supporting the role of proton-dependent oxide-mediated pathways in surface degradation. 159 Electrochemical quartz crystal microbalance studies by Sakurai et al. also indicate that hydroxylated Pt intermediates, such as Pt(OH)3+, are central to the dissolution mechanism in HClO4 and H2SO4. 157 These studies illustrate that while pH and oxide-mediated pathways govern Pt dissolution in nonchloride acidic media, the extent of actual metal solubilization remains limited.
Effects of potential and scan rate on Pt and Ir dissolution
Regarding the relationship between applied potential and dissolution of Pt, it has been reported that the UPLs significantly impact the amount of Pt dissolved, with higher UPLs leading to increased concentrations of dissolved Pt.75,160 At lower UPLs, such as 0.85 V versus RHE, only trace amounts of Pt were detected, indicating reduced platinum dissolution under these conditions.131,161 Jovanovic et al. and Topalov et al. further concluded that significant Pt dissolution occurs only when the UPL during cycling reaches ≥1.1 V versus RHE, and that dissolution becomes dominant at UPLs ≥1.3 V versus RHE during the negative-going potential sweep.91,145,162,163 Cherevko et al. observed the onset dissolution potential at 1.05–1.1 V versus RHE for Pt in 0.1 M HClO4. 75 Furthermore, Furuya et al. reported that the amount of Pt dissolved during potential cycling is greater than that dissolved during potential holding.145,163 Overall, the primary platinum dissolution process in aqueous electrolytes has been reported to be cathodic dissolution, occurring during the reduction of surface oxides at potentials greater than 1.2 V versus RHE. 101
It was shown that the dissolution of hydrous iridium oxide depends on the potential of the electrode as well as the thickness of the oxide layer, with increases in these parameters leading to enhanced dissolution.111,112 While iridium is generally inert in acidic solution under static conditions, it dissolves at a measurable rate during potential cycling. 164 The dissolution behavior of the iridium electrode was monitored by cycling between 0.06 and 1.54 V versus RHE in 1 M H2SO4 for 3000 cycles at a scan rate of 40 mV/s. Jovanovic et al. conducted a dissolution study on an iridium-based electrode using a thin-film rotating disk electrode, cycling the potential between 1.00 and 1.63 V versus SCE at a scan rate of 10 mV/s for 10,000 cycles. ICP-MS analysis and electrochemical dissolution profiles revealed that iridium dissolution occurs well below the typical OER potentials. 138
Pt is reported to be electrochemically stable in acidic environments at low potentials, but undergoes dissolution at high potentials in acidic environments.
165
The potential Eh–pH diagram in Figure 8 illustrates the thermodynamic behavior of Pt as a function of electrode potential and pH, indicating its stable phase and the oxidation state.166,167 According to this diagram, Pt oxide (PtO
x
) is formed at high potentials under acidic conditions, leading to dissolution.
168
Reaction (7) represents the oxidation of metallic Pt to its divalent cation Pt2+, a process influenced by the surface structure of Pt and the presence of oxygen. This transformation occurs in acidic environments at potentials where Pt is oxidized but below the onset of bulk oxide formation.
71
Reaction (8) represents the formation of platinum monoxide (PtO) from metallic Pt in the presence of water. This process is facilitated by the adsorption of hydroxyl groups on the Pt surface. The formation of PtO typically occurs at higher potentials than the formation of Pt2+ but remains within the stability range of PtO.
45
Reaction (9) represents the further oxidation of platinum monoxide (PtO) to platinum dioxide (PtO2) in the presence of water. PtO2 is more stable at higher potentials than PtO and typically forms under stronger oxidative conditions.167,169
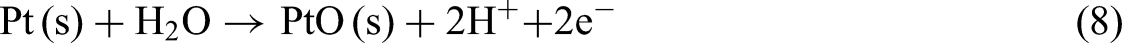
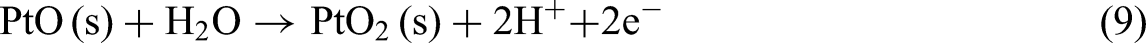
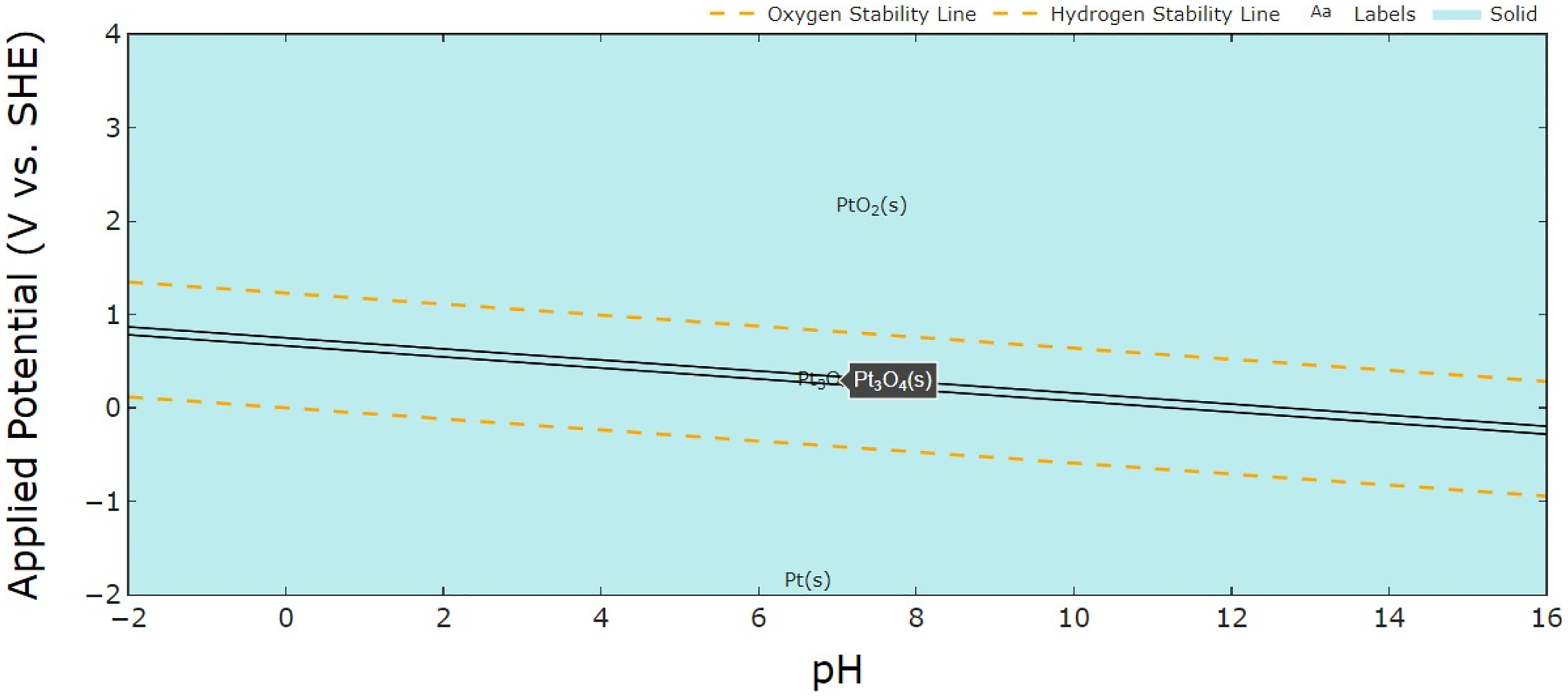
The potential Eh–pH diagram of platinum. The dashed lines separate the stability regions of water, while the solid lines indicate the stability regions of different Pt species. The blue area represents the solid phase.
Iridium is considered a precious metal due to its exceptional thermal and physical properties, as well as its limited natural abundance (Figure 9). The increasing industrial demand for iridium has prompted researchers to develop effective recovery methods for recycling iridium from secondary resources.170–173 Iridium is one of the most corrosion-resistant of all the PGMs, which makes it difficult to recycle. However, certain electrochemical conditions allow for its dissolution. Wang et al. reported that at pH = 0, IrO2 transforms to IrO3 at the potential of 1.55 V, and IrO3 dissolves to IrO4− at the potential of 1.60 V.
174
Reaction (10) describes the oxidation of metallic iridium (Ir) to the Ir3+ ionic form, which is significantly influenced by pH levels and the electrochemical environment. This transformation is particularly relevant under acidic conditions where Ir3+ can remain stable, especially during the OER.46,48 Reaction (11) represents the oxidation of metallic Ir to IrO2 in the presence of water. The stability region of IrO2 is typically at higher potentials, as indicated in the Pourbaix diagram.
49
Reaction (12) illustrates the further oxidation of IrO2 to the soluble species 
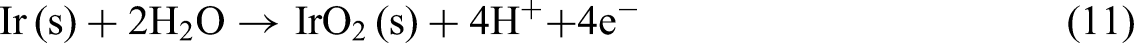
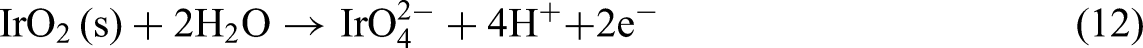
The potential Eh–pH diagram of iridium. The dashed lines separate the stability regions of water, while the solid lines indicate the stability regions of different Ir species. The blue area represents the solid state, and the white area represents the ion state.
Recent advances in electrochemical PGM recovery
Electrochemical dissolution of electrocatalysts has emerged as a promising alternative for recycling PGMs from secondary resources, such as spent PEM MEAs. 167 Conventional PGMs recover from primary ores in mining involve complex hydro-pyrometallurgy processes that are both expensive and hazardous. On the other hand, recovering PGMs from secondary resources is more cost-effective and practical, as secondary resources possess higher concentrations of PGM than their corresponding ore. These materials not only offer a richer PGM content but also reduce the environmental impact and energy consumption associated with primary mining and extraction. With the increasing demand for high-purity PGMs, developing efficient and environmentally friendly processes has become a priority.176,177 Research suggests that conventional recovery methods, which often require harsh chemical conditions, can be effectively replaced by electrochemical techniques. These novel methods offer safer, cost-effective, and highly efficient recovery while enabling the extraction of additional valuable materials. Figure 10 illustrates a novel electrochemical process developed by Latsuzbaia et al. for recovering both the electrocatalyst and the carbon support from MEAs. This selective recovery process demonstrates high potential for reuse as it is fast, has low operational complexity, enables the recovery of multiple materials and offers high recovery efficiency.7,83
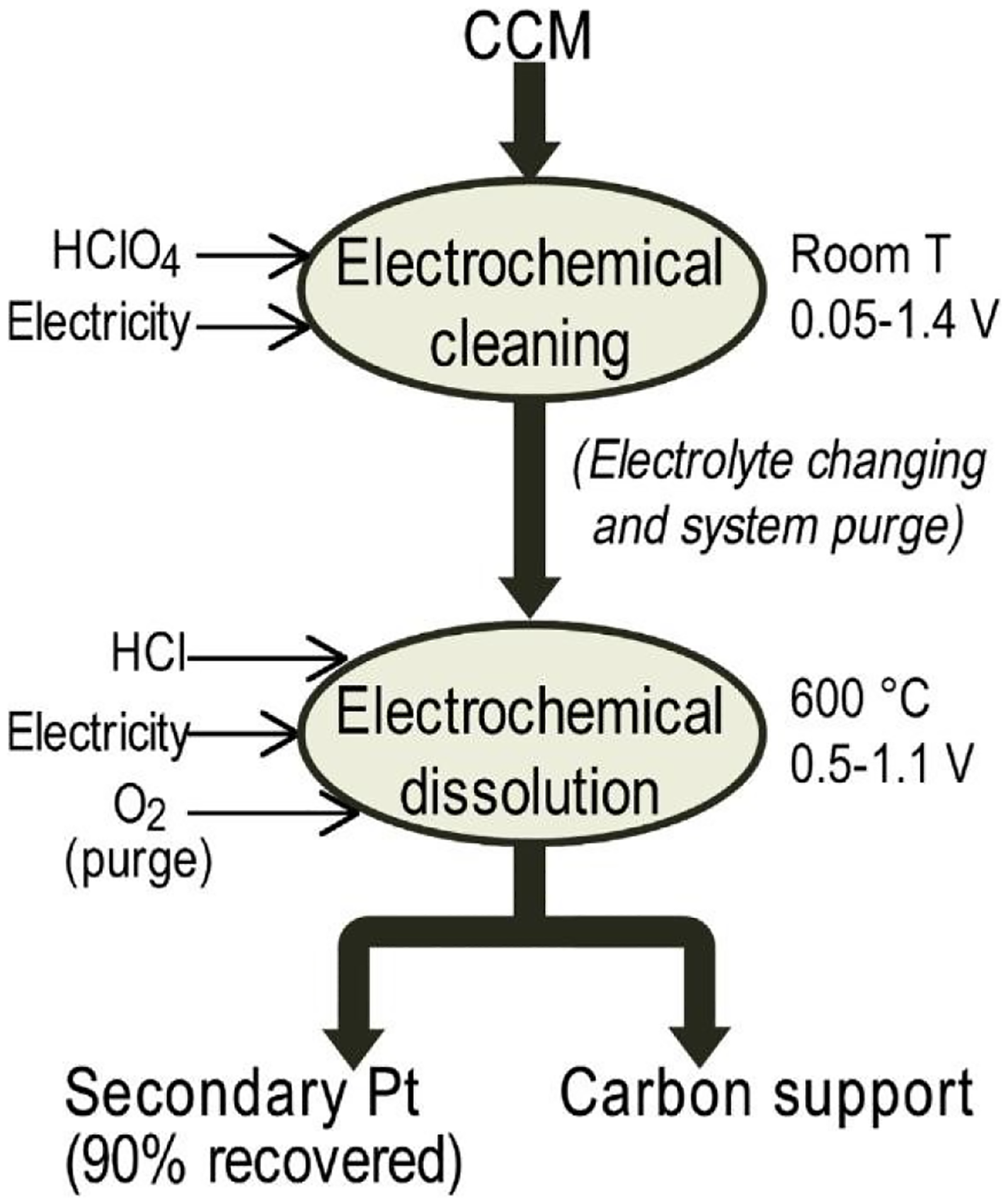
Electrochemical dissolution of Pt and carbon support recovery. 7 (Reproduced with permission from Antonio Valente, Diego Iribarren, and Javier Dufour, under license (https://creativecommons.org/licenses/by-nc-nd/4.0/)).
Challenges and future perspectives
Electrochemical dissolution studies have significantly advanced the understanding of PGM degradation mechanisms and have enhanced the recovery of these valuable metals through selective and controlled processes, offering clear advantages over conventional recovery methods. However, several challenges remain. One major issue is that the dissolution of Pt and IrO x is a kinetically slow process, often requiring prolonged durations or the application of high voltages. This poses a problem, as ongoing research efforts are focused on minimizing high-voltage operations due to their high energy consumption and environmental impact. To address this, future work should further focus on optimizing key dissolution parameters such as temperature, potential window, and electrolyte composition to accelerate dissolution rates without the need for elevated voltages. Another significant challenge is the recovery of electrocatalysts in high-purity salt form with controlled morphology. The presence of impurities and mixed metal ions from MEAs interferes with the selective isolation of the desired metals. Future research should focus on integrating electrochemical dissolution with additional purification steps, such as the use of ion-exchange membranes to improve the separation and purity of recovered Pt and Ir salts. Additionally, undesirable electrochemical processes such as electrocatalyst redeposition, carbon corrosion, and particle agglomeration can interfere with dissolution outcomes, leading to reduced recovery yields. To mitigate their impact, future research should focus on understanding the underlying mechanisms of these side reactions to enable better control of the electrochemical environment.
Conclusion
The recovery of PGMs from secondary resources has been investigated through various approaches, each offering distinct benefits and limitations. While hydrometallurgical processes are recognized for their lower energy consumption and higher selectivity compared to pyrometallurgical processes, they also pose significant environmental concerns due to the large quantities of hazardous and toxic waste generated. This highlights the urgent need for more sustainable and environmentally friendly recovery methods. In the context of hydrogen fuel cells, the degradation of electrocatalysts, specifically during transient dissolution under oxidation and reduction processes, is a key factor contributing to the reduced lifespan of these systems. This has led researchers to focus on electrochemical dissolution as a more environmentally and economically viable EoL method for recovering anode- and cathode-deposited electrocatalysts.
Among the electrochemical techniques reviewed, potential cycling and square wave potential cycling emerge as promising methods for the effective recovery of spent electrocatalysts. Potential cycling has been shown to promote platinum dissolution at potentials ranging from 0.6 to 1.2 V versus RHE, with dissolution rates increasing at higher potentials. Similarly, iridium dissolution is enhanced within the potential range of 0.06 to 1.54 V versus RHE. Notably, square wave potential cycling has demonstrated enhanced dissolution rates for both Pt and Ir when compared to potential cycling, making it a promising approach for maximum recovery of electrocatalysts. The review also discussed the influence of scan rate on the dissolution of Pt and Ir from the Pt/C and IrO2 electrocatalysts. The role of chloride (Cl−) anions in enhancing dissolution rates has also been highlighted, with HCl electrolytes proving more effective in promoting electrocatalyst dissolution than HClO4 and H2SO4 electrolytes. Overall, the reviewed electrochemical strategies contribute toward achieving sustainable development goals, which ensures affordable, reliable, and sustainable clean energy. The reviewed technology gives a platform to scale PGM supply through safe, sustainable, and efficient recycling.
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Research Foundation (grant number 138079).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.