
Abstract
Future fuels for compression ignition engines will be required both to reduce the anthropogenic carbon dioxide emissions from fossil sources and to contribute to the reductions in the exhaust levels of pollutants, such as nitrogen oxides and particulate matter. Via various processes of biological, chemical and physical conversion, feedstocks such as lignocellulosic biomass and photosynthetic micro-organisms will yield a wide variety of potential fuel molecules. Furthermore, modification of the production processes may allow the targeted manufacture of fuels of specific molecular structure. This paper therefore presents an overview of the effects of fuel molecular structure on the combustion and emissions characteristics of compression ignition engines, highlighting in particular the submolecular features common to a variety of potential fuels. An increase in the straight-chain length of the alkyl moiety reduces the duration of ignition delay, and the introduction of double bonds or branching to an alkyl moiety both increase ignition delay. The movement of a double bond towards the centre of an alkyl chain, or the addition of oxygen to a molecule, can both increase and decrease the duration of ignition delay dependent on the overall fuel structure. Nitrogen oxide emissions are primarily influenced by the duration of fuel ignition delay, but in the case of hydrogen and methane pilot-ignited premixed combustion arise only at flame temperatures sufficiently high for thermal production. An increase in aromatic ring number and physical properties such as the fuel boiling point increase particulate matter emissions at constant combustion phasing.
Keywords
Introduction
International recognition as to the devastating impacts of anthropogenic emissions of greenhouse gases (GHGs) on global climate change 1 has led to the requirement that future use of internal-combustion (IC) engines should result in reduced emissions of carbon dioxide (CO2) from fossil sources relative to current levels. Concurrent to this has been the introduction of increasingly stringent legislation 2 regulating levels of toxic pollutants, such as nitrogen oxides (NO x ) and particulate matter (PM) from road transport; however, a relatively unexplored opportunity for the potentially simultaneous control of various harmful pollutants (GHGs, NO x and PM) 3 from IC engines is by the molecular design of future fuels.
Over the course of the past 100 years in which IC engines have been in widespread use, many improvements have been made to the mechanical design and combustion strategies utilised, resulting in significantly higher engine thermal efficiencies (and thus reduced specific emissions of CO2).4,5 Simultaneously, exhaust after-treatment devices are now employed in both spark ignition engines and compression ignition engines to ensure compliance with emissions regulations and to reduce the exhaust levels of carbon monoxide, unburned hydrocarbons, NO x and PM. However, the use of such devices can result in increased fuel consumption and reduced engine thermal efficiency as they require a sufficiently high exhaust temperature for proper operation.4,6
The composition of fuels for IC engines has, however, remained relatively unchanged (with the exceptions of the removal of sulphur and the varying use of additives) and continues to be specified by the physical properties, e.g. the boiling-point range, and combustion regime specific measures such as cetane number in the case of fuels for compression ignition.7,8 The most significant impact in recent years on the composition of fuels for IC engines within EU member states has been the regulatory requirement that biofuels must comprise a mandatory 10% minimum of gasoline and diesel consumption for road transport by 2020, as stipulated by the 2009 European Parliament Directive on the use of energy from renewable sources. 9 In the case of fuels for compression ignition engines, it has been recognised that biodiesel, which is the most commonly utilised renewable fuel component, exhibits combustion and emissions characteristics different from those of conventional fossil diesels.10–16 However, attempts to design a fuel composition for improved engine efficiency or reduction of exhaust pollutants have not been widely reported, despite the opportunities to do so presented by the legislative need to develop renewable fuels from non-food sources.
Current liquid fuels from fossil sources are typically mixtures of straight-chain and branched-chain (iso)alkanes and (iso)alkenes, cyclic and aromatic molecules. Table 1 shows the molecular structures of typical hydrocarbon molecules contained within fossil diesel and also those of potential alternative fuel molecules from renewable sources. Although much focus has been placed on the identification of non-edible feedstocks for the production of biodiesel, 17 a wide range of chemical and biological processes are currently under study with potential for the production of fuel molecules suitable for compression ignition combustion. Alkanes, alkenes, alcohols, methane and hydrogen can be produced from genetically modified micro-organisms including photosynthetic micro-algae,18–21 whereas catalytic upgrading of fermentation products can result in higher-weight alcohols and ketones.22,23 Many methods of combined thermal, chemical and biological treatment of lignocellulosic biomass can produce ketones, short-chain fatty acid esters, acids, furans, methane and phenols.24–30
Molecular structures of typical current and future fuels for compression ignition engines.
With the development of various controlled processes for the conversion of renewable energy sources to liquid fuels for compression ignition engines, in contrast with current fossil diesel, at the point of production these future fuels will likely consist of a relatively small and discrete range of species, thereby enhancing the importance of the molecular structures of the compounds present to the overall fuel characteristics. This paper therefore presents a concise review of the effects of the molecular structures of fuels on the combustion characteristics and the exhaust emissions of NO x and PM in modern direct-injection compression ignition engines, focusing in particular on the influence of the alkyl chains which are common to many potential fuel molecules.
Effects of the molecular structure of fuels on the duration of ignition delay
Alkyl chain length and branching
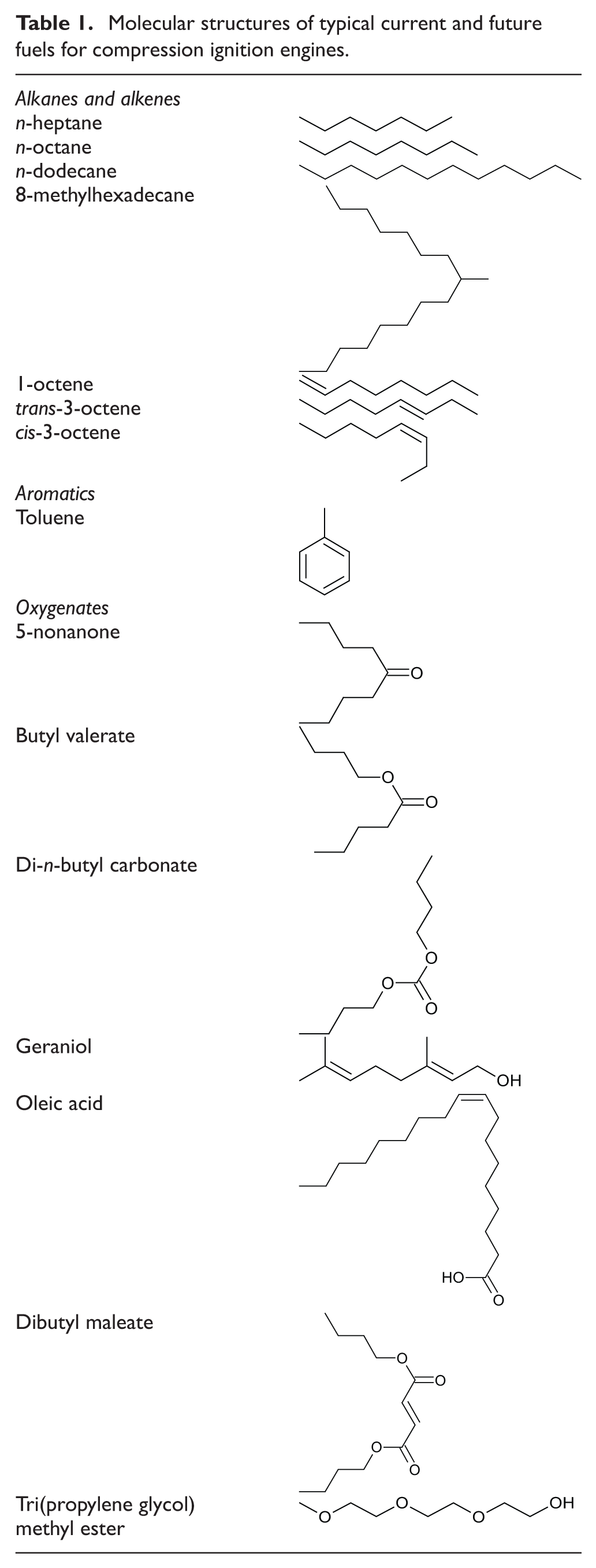
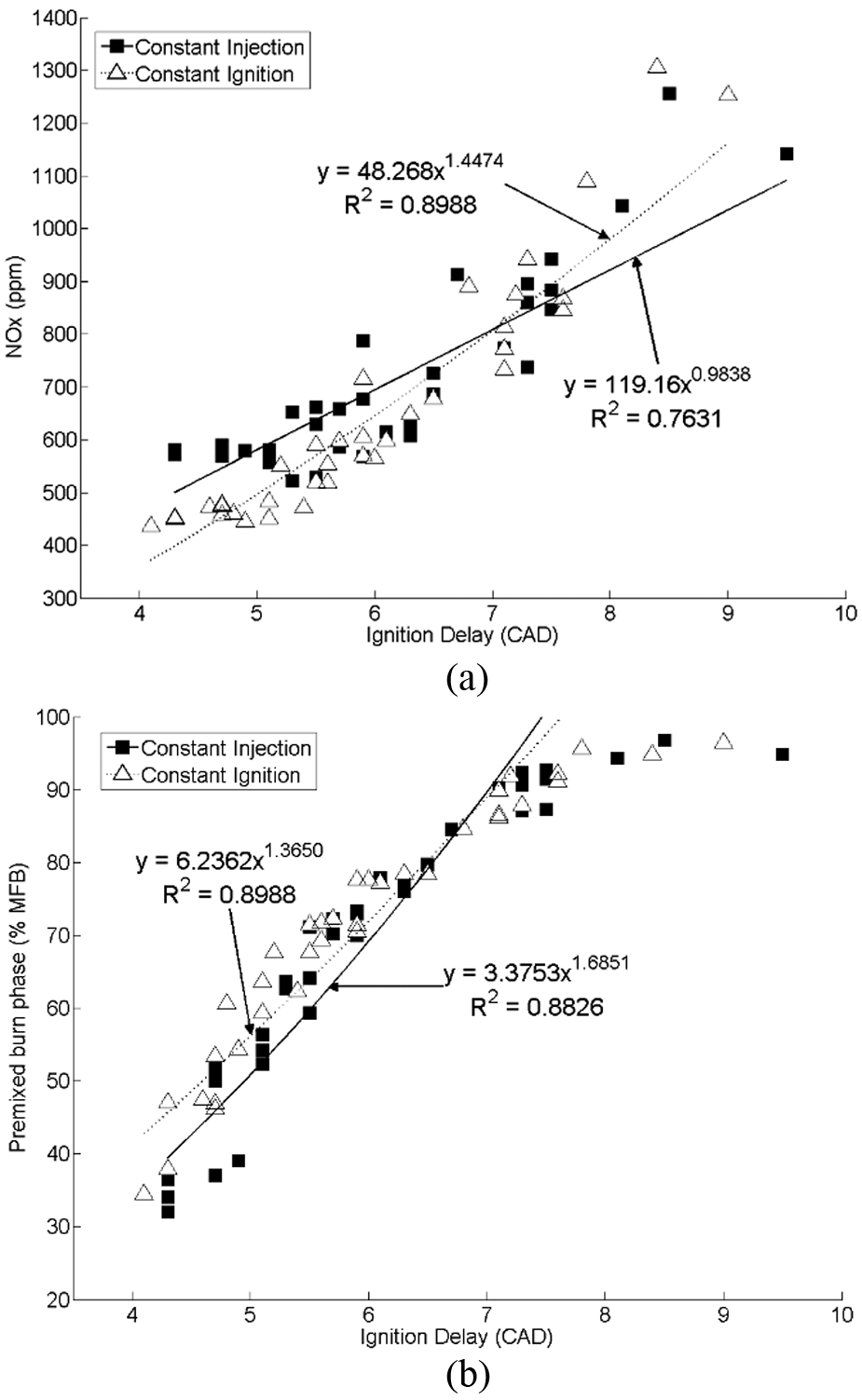
Many of the hydrocarbon and oxygenate molecules which constitute fossil fuels, current biofuels and future non-fossil alternatives (Table 1), contain at least one alkyl chain which will typically account for the majority of the molecular mass. Figure 1 shows the impact of increasing the n-alkane alkyl chain length on the duration of ignition delay, defined as the interval between the start of fuel injection (SOI) and the start of combustion (SOC), when tested as single-component fuels at a constant engine speed of 1200 r/min, a fuel injection pressure of 450 bar and a constant engine work output (indicated mean effective pressure (IMEP)) of 4 bar 31 The fuels were tested at two injection timings: first, constant injection at which the SOI was held constant and the SOC was allowed to vary in accordance with the ignition delay of the fuel; second, constant ignition at which the SOC was fixed at top dead centre and the SOI was varied with the duration of the fuel ignition delay. Figure 1 shows that, as the n-alkane alkyl chain length is increased, there is a consistent decrease in the duration of ignition delay at both injection timings. It can also be seen in Figure 1 that the addition of carbon atoms to the alkyl chain is more significant in the case of shorter-chain n-alkanes. For example, the addition of four carbon atoms to increase the alkyl chain length from eight atoms to 12 atoms (n-octane to n-dodecane) decreases the ignition delay by approximately 1° crank angle (CA), whereas the addition of four carbon atoms to increase the alkyl chain length from 16 atoms to 20 atoms (n-hexadecane to n-eicosane) decreases the ignition delay by about 0.5° CA. The observed diminishing decrease in ignition delay with increasing alkyl chain length concurs with measurements of the cetane numbers of n-alkanes, 32 and also kinetic modelling simulations of shock tube ignition delay times of C8 to C16 n-alkanes in conditions relevant to compression ignition engines. 33 The results of these experimental investigations and modelling simulations are in agreement with the effect of alkyl chain length on rates of low-temperature radical branching reactions during the ignition delay period. 34 The progress of such reactions determines the rate at which radical species (primarily H2O2) accumulate and local temperatures escalate towards those required for the rapid decomposition of radicals and subsequent fuel autoignition.
Effect of increasing n-alkane alkyl chain length on the ignition delay at constant fuel injection timing and constant SOC timing. Source: reproduced with permission from SAE International, 2011. 31
Figure 2 shows the low-temperature reaction steps of hydrogen abstraction, oxygen addition and fuel molecule isomerisation required for the propagation of radical species from n-octane and 2-methylpentane. Rates of hydrogen abstraction increase with increasing alkyl chain length, as this results in the availability of more secondary carbon atoms (carbon atoms 2 to 7 in Figure 2(a)) which possess a lower carbon to hydrogen bond strength than primary carbon atoms (carbon atoms 1 and 8 in Figure 2(a)). 34 An increase in the alkyl chain length also increases the rates of isomerisation following oxygen addition as a greater number of sites at which six-member or seven-member transition rings are able to form become available, as this requires chains of at least three fully saturated carbon atoms. 35
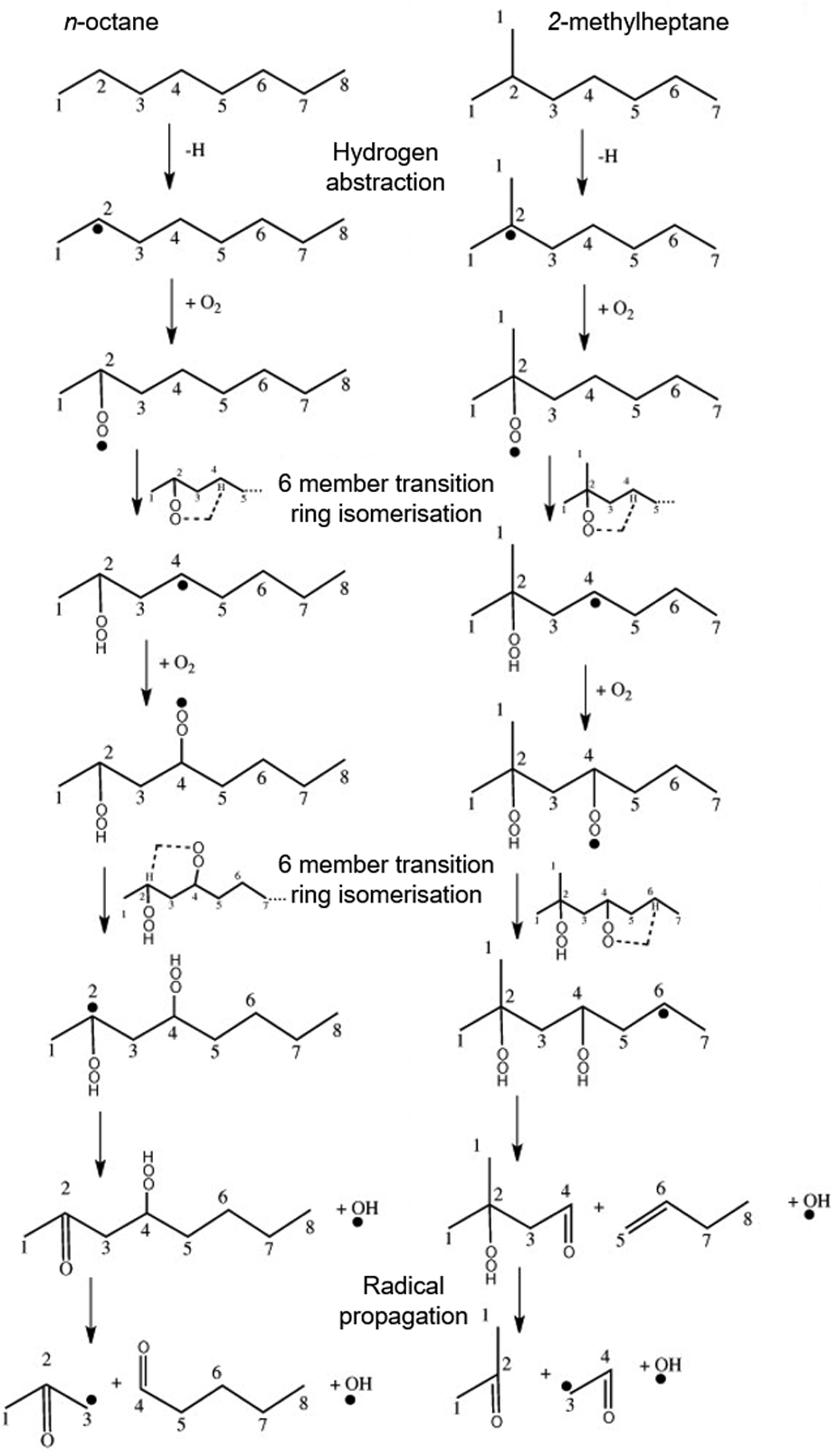
Initial low-temperature branching reactions of (a) n-octane and (b) 2-methylpentane.
A decrease in ignition delay with increasing alkyl chain length has also been observed in molecules other than n-alkanes.32,36,37 For example, Schönborn et al. 36 and Koivisto et al. 37 found a linear decrease in the duration of ignition delay with increasing alkyl chain length in the case of fully saturated fatty acid methyl esters (FAMEs) with alkyl chain lengths of between 12 carbon atoms to 22 carbon atoms, and primary and secondary alcohols with an alkyl chain length of between 8 carbon atoms to 16 carbon atoms, respectively. However, it is interesting to note that, in the case of fatty acid esters produced from alcohols other than methanol (and thus possessing an additional alkyl chain within the alcohol moiety of the ester), the influence of the alkyl chain length of the alcohol moiety on the duration of ignition delay is insignificant relative to that of a change in the alkyl chain length of the fatty acid moiety.36,38–41
The presence of alkyl chain methyl branches in n-alkanes31,31 (and also in the alkyl chains of other molecules such as ethers 42 and carbonate esters 43 ) has been observed to result in an increase in the duration of ignition delay. In engine tests of 8-methylhexadecane and 2,6,10,16-tetramethylpentadecane, the increase in ignition delay was equivalent to, or more significant, than that observed when comparing the n-alkane of equal carbon number to the branched alkane to the n-alkane of equal straight alkyl chain length as the iso-alkane. 31 In Figure 2(b), it can be seen that, whereas the low-temperature branching reactions of 2-methylpentane initiate with hydrogen abstraction from a tertiary carbon atom (the bond strength of which is weaker than those of secondary and primary carbon to hydrogen bonds), the second isomerisation differs from that in the case of n-octane as the carbon atom bonded to the first OOH moiety is not included in the second six-member transition ring formed (Figure 2(a)). This is because there are no further hydrogen atoms available for abstraction from this tertiary carbon atom in the case of 2-methylpentane, and the rate of the second isomerisation is therefore slower than that in an n-octane-derived radical as hydrogen abstraction from a secondary carbon atom is required. 33
Degree of unsaturation and positions of double bonds in an alkyl chain
Figure 3 shows the change in ignition delay with the percentage of double carbon-to-carbon bonds present within the total carbon chain length at a constant injection timing of 7.5° CA before top dead centre and at the same constant engine operating conditions as the results presented in Figure 1. 31 It can be seen in Figure 3 that there is a linear increase in ignition delay with increasing degree of chain unsaturation proportional to the percentage of double bonds present, irrespective of the overall carbon chain length. The presence of double bonds within carbon chains decreases the rates of low-temperature reactions (Figure 2), by reducing both the number of secondary hydrogen atoms available for abstraction and the number of sites at which six-member transition rings can form. 34
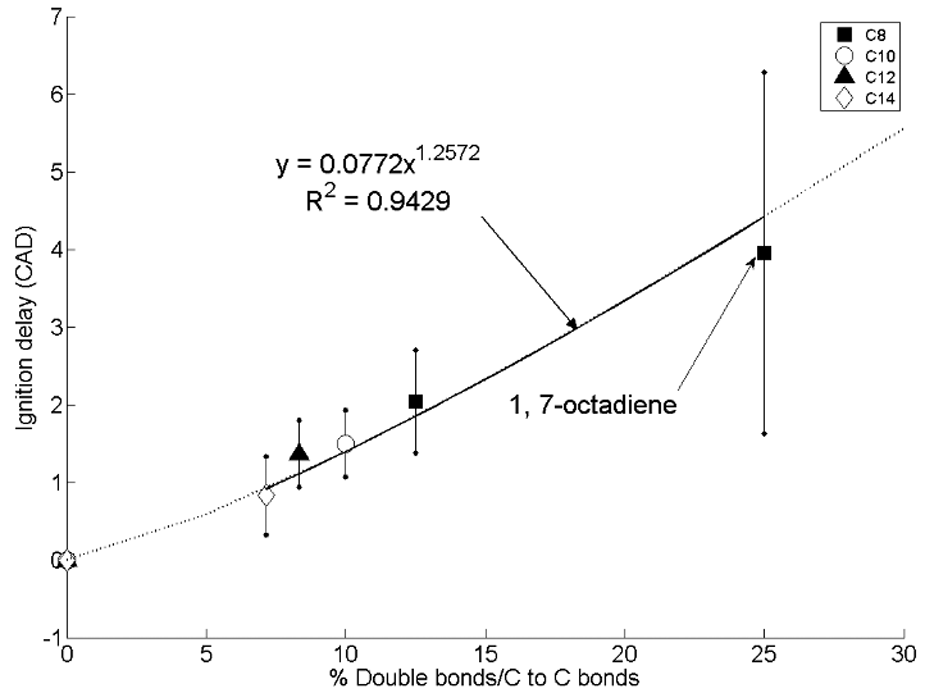
Change in the duration of ignition delay with the percentage of carbon-to-carbon double bonds present in C8 to C14 n-alkanes and straight-chain alkenes.
Figure 4 shows the ignition delays of n-octane, 1-octene, trans-2-octene, cis-3-octene and trans-3-octene relative to a reference fossil diesel in a direct-injection diesel engine at constant injection timing and constant ignition timing. 44 It can be seen in Figure 4 that, at both injection timings, moving the double-bond position of octene from position 1 to position 2 results in an increase in the duration of ignition delay; however, further movement of the double bond towards the centre of the molecule reduces the ignition delay, in the case of both cis-3-octene and trans-3-octene. This observation is contrary to studies of shorter-chain alkene isomers in rapid compression machines; Tanaka et al. 45 and Vanhove et al. 46 found that moving the position of the double bond progressively towards the centre of a molecule consistently resulted in an increased duration of ignition delay in heptene and hexene trans isomers. Zhang et al. 47 found a similar influence of the double-bond position within the alkyl chain of a fatty acid ester in motored premixed combustion in a Cooperative Fuels Research engine, with the shortest duration of ignition delay exhibited by methyl nonanoate, followed by methyl trans-2-nonenoate, and with the longest ignition delay observed in the case of methyl trans-3-nonenoate.
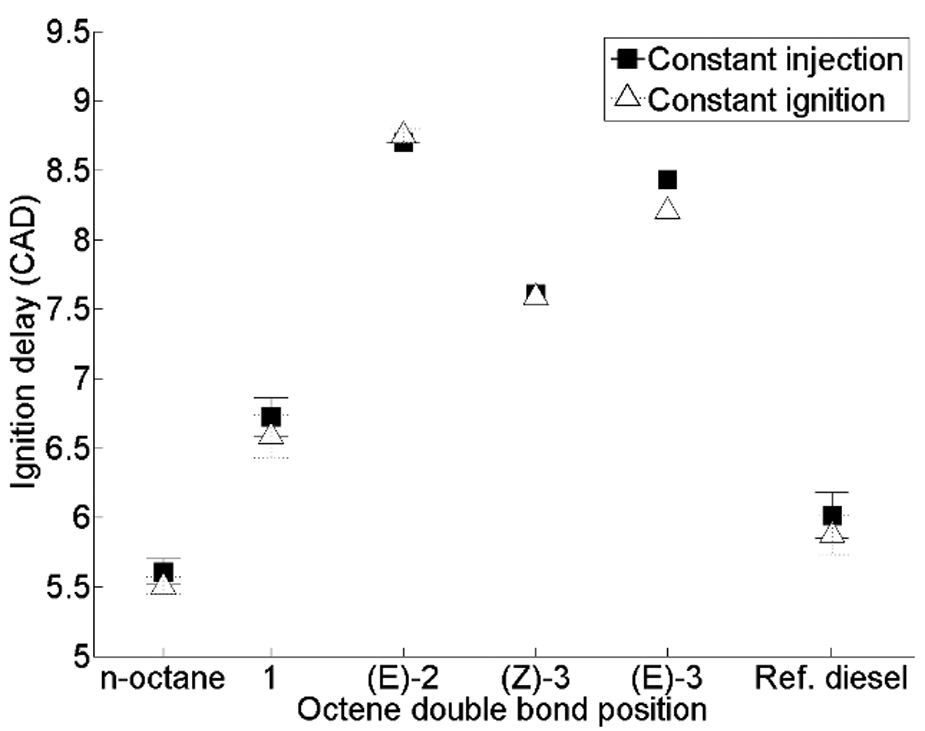
Ignition delay of octene isomers, n-octane and reference diesel at constant injection timimg and constant ignition timing.
Fridlyand et al. 48 investigated the relative reactivities (using speciation of stable intermediates by gas chromatography) of 1-decene, cis-2-decene, cis-5-decene and trans-5-decene in a shock tube at conditions (40–66 bar and 850–1500 K) relevant to compression ignition engines. Contrary to studies of shorter-chain alkene isomers,44–46 reactivity was found to increase with the movement of the double bond towards the centre of the molecule. 48 In the case of hexene isomers, it has been suggested that the decrease in reactivity with the movement of the double bond can be attributed to the decreasing length of the saturated alkyl chains within the molecule, and the concurrent increasing importance of low-temperature reactions involving the double bond.46,49 Hellier et al. 44 suggested that the initial increase in the duration of ignition delay (Figure 4) in engine tests of octene isomers can be similarly explained by the decreasing length of the residual saturated alkyl chain but that, in moving the double bond from position 2 to position 3, the longer saturated chain lengths relative to hexane and heptane isomers resulted in a net increase in the low-temperature reactivity. Kinetic modelling undertaken by Fridlyand et al. 48 suggested that decene isomers undergo significantly different reaction pathways dependent on the double-bond position and, in a further study investigating methyl trans-2-nonenoate, methyl trans-3-nonenoate and the analogous alkenes, 1-octene and trans-2-octene, found a dissimilar influence of double-bond movement within the alkyl chain in the FAMEs relative to the alkenes. 50
Figure 4 also shows a reduced duration of ignition delay of cis-3-octene relative to that of trans-3-octene, and this was suggested to have been due to the requirement for alkenyl and alkenyl peroxy radicals to be in the cis arrangement prior to internal isomerisation across the double bond during low-temperature branching reactions.44,51 However, in the shock tube study of decene isomers by Fridlyand et al., 48 no discernible difference between the reactivities of cis-5-decene and trans-5-decene was detected, further highlighting the varying influence of the double-bond position and arrangement with the overall molecular structure.
Oxygen-bearing functional groups: esters, alcohols, ketones and ethers
Figure 5 shows the effects on the duration of ignition delay of adding oxygen to a molecule of constant carbon number in the case of the carbonyl group 43 and as ether linkages. 52 In Figure 5(a), it can be seen that adding an oxygen atom to 5-nonanone to form butyl valerate increases the duration of ignition delay, as does the addition of a further oxygen atom to form di-n-butyl carbonate. Conversely, it can be seen in Figure 5(b) that the addition of oxygen atoms within the alkyl chain as ether linkages results in a decrease in ignition delay. In both cases (Figure 5(a) and (b)), the effects of increasing the molar oxygen content have been attributed to changes in the rates of hydrogen abstraction and internal isomerisation during low-temperature branching reactions (Figure 2). The increased oxygen content of a fatty acid ester relative to a ketone (and of a carbonate ester relative to a fatty acid ester) decreases the rate of initial hydrogen abstraction, because of the increasing electronegativity of the functional group, 25 and also decreases the number of alkyloxy radicals formed as the presence of an oxygen atom attached to the alkyl chain decreases the rates of transition ring reactions.43,53 However, the presence of oxygen atoms within the alkyl chains of ethers increases the rate of hydrogen abstraction (by reduction in the adjacent C–H bond strength), the rates of isomerisation and also subsequent radical propagation.52,54–56
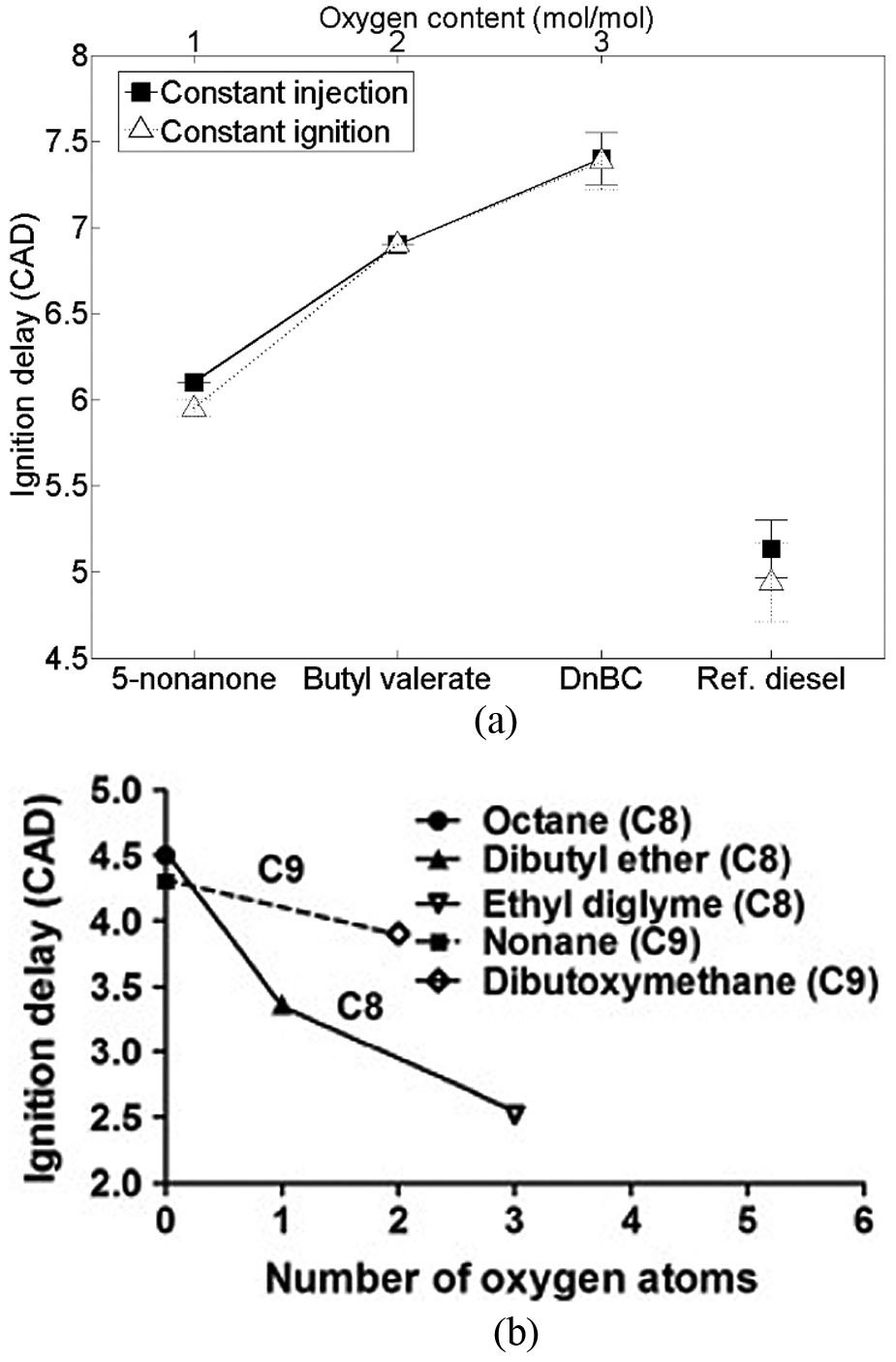
Effect of the molecular oxygen content on ignition delay of (a) di-n-butyl carbonate, butyl valerate and 5-nonanone and (b) C8 and C9 ethers.
By studying various terpenes potentially produced by genetically modified cyanobacteria, Hellier et al. 57 observed that, of the molecules tested which represented a change to only the functional group of geraniol (a C10 alcohol), the equivalent aldehyde, geranial, exhibited the largest decrease in the duration of ignition delay. This is consistent with cetane number measurements of aldehydes relative to equivalent alcohols (octanal and octanol 32 ); aldehydes are known to be intermediates in the low-temperature reactions of alcohols, 58 the higher reactivity of which can be attributed to the relative ease with which peroxy radicals may be formed, requiring the abstraction of a single hydrogen atom without the addition of oxygen. 59
Many studies have observed an increase in the ignition delay of molecules containing oxygenated functional groups in the terminal position relative to those of alkanes or alkenes of equivalent alkyl chain length.40,52,53 However, in the study of terpenes by Hellier et al., 57 where the alkyl moiety present was both branched and unsaturated, the equivalent alkene, citronellene, was of significantly poorer ignition quality than those of the various oxygenated molecules tested.
Binary mixtures including aromatic molecules
Figure 6 shows the duration of ignition delay of binary toluene–n-heptane and 1-octene–n-octane mixtures relative to a reference fossil diesel at constant injection timing and constant ignition timing. In Figure 6(a), it can be seen that increasing the percentage of toluene present in the toluene–n-heptane binary mixtures from 0% to 30% results in a near-linear increase in ignition delay for both timings, and while at toluene levels greater than 35% toluene there continues to be a linear increase in ignition delay, the gradient of this relationship increases significantly. 60 The ignition retarding effect of toluene addition has also been observed in both homogeneous charge compression ignition engines 61 and Cooperative Fuels Research engines, 62 and in the study of alkylbenzenes by Koivisto et al., 63 where binary toluene–n-alkane mixtures were found to have longer durations of ignition delay than the alkylbenzene of corresponding alkyl chain length. Figure 6(b) shows that increasing the percentage of 1-octene present in 1-octene–n-octane mixtures also results in a linear increase in the duration of ignition delay; however, in contrast with the toluene–n-heptane mixtures, the gradient of this relationship remains almost constant over the whole range of 1-octene additions. 60
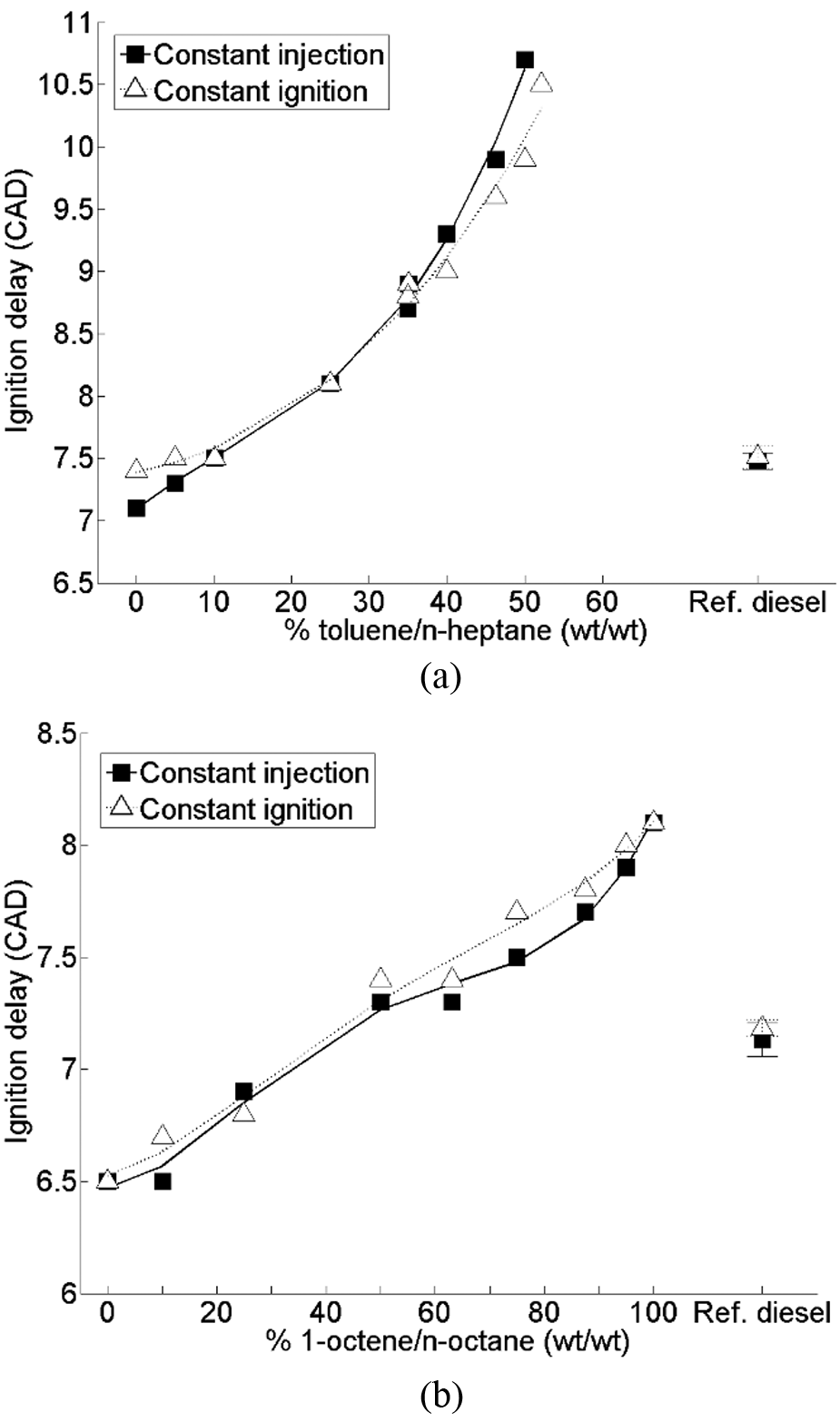
Durations of ignition delay of (a) toluene–n-heptane mixtures and (b) 1-octene–n-octane mixtures at constant injection timing and constant ignition delay timing.
The non-linear effect of toluene addition on the duration of ignition delay has been suggested to be attributable, in both premixed combustion systems61,64 and direct-injection compression ignition combustion 60 (Figure 6(a)), to the inhibiting effect of toluene on the low-temperature branching reactions of n-alkanes. At low temperatures, toluene can be oxidised via the abstraction of a hydrogen atom from the methyl group by oxygen or preferentially non-selective OH radicals, resulting in benzyl radicals that are thermally stable below 1000 K and cannot be further oxidised.59,61,64 This consumption of an OH radical into a stable benzyl species therefore slows the rates of n-alkane branching reactions, in which the radical would otherwise be available to participate.
Premixed hydrogen–methane pilot-ignited combustion
Figure 7 shows the variation in the duration of ignition delay period for hydrogen–diesel (H2–diesel) fuel co-combustion tests carried out by Talibi et al. 65 on a naturally aspirated direct-injection diesel engine. Although hydrogen flames are expected to have high propagation rates owing to the fast and thermally neutral branching-chain reactions of hydrogen relative to the slower endothermic low-temperature reactions of larger fuel molecules, 66 hydrogen has an appreciably lower cetane number than that of fossil diesel fuel 66 and cannot be readily ignited by means of compression in modern diesel engines. Therefore, for the experiments presented in Figure 7, the duration of ignition delay is that of the diesel fuel, the combustion of which resulted in ignition and flame propagation through the intake-air-aspirated hydrogen.
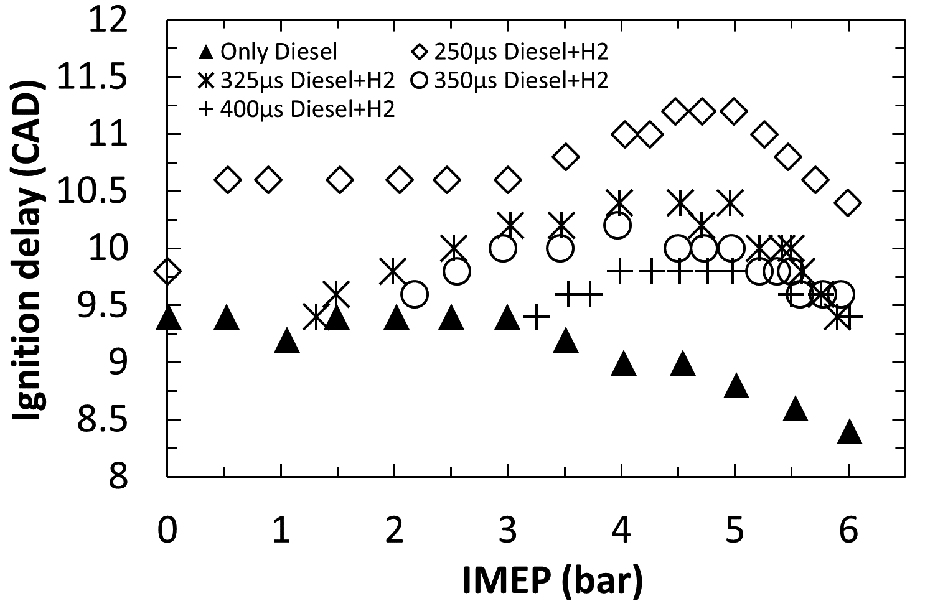
Durations of ignition delay for H2–diesel co-combustion experiments carried out at a fixed direct-injection diesel fuel flow rate and an increasing level of intake air-aspirated hydrogen addition to increase the engine load.
Figure 7 shows that for each set of tests, as the engine load is increased (by hydrogen addition, while the diesel fuel flow is kept constant), there is a general trend of increasing ignition delay, which peaks and subsequently decreases with further hydrogen addition. The initial increase was attributed to the displacement of intake oxygen by hydrogen, which led to a decrease in the rates of low-temperature fuel breakdown reactions (Figure 2), thereby delaying autoignition. At higher engine loads (and thus higher levels of hydrogen addition and a subsequently richer hydrogen–air (H2–air) stoichiometry) it was suggested that the reduction in ignition delay arises from a higher in-cylinder temperature which offsets the reduced oxygen level. 65
Figure 8 shows similar trends in ignition delay when burning methane–hydrogen (CH4–H2) mixtures, pilot ignited by diesel fuel, in a compression ignition engine 67 as seen in the case of hydrogen only intake aspirated mixtures (Figure 7). Furthermore, Figure 8 shows that, for the same engine load, the ignition delay period is considerably longer for all CH4–H2 mixtures relative to only diesel fuelling. Whereas diesel fuel is expected to burn at near-stoichiometric conditions, 68 the aspirated CH4–H2 mixture is relatively lean and cannot be expected to participate chemically in the diesel fuel low-temperature reactions during the ignition delay period (Figure 2). It was also speculated that the aspirated CH4–H2 mixture slows oxidation reactions by diluting the intake oxygen, resulting in an increased ignition delay period. 67
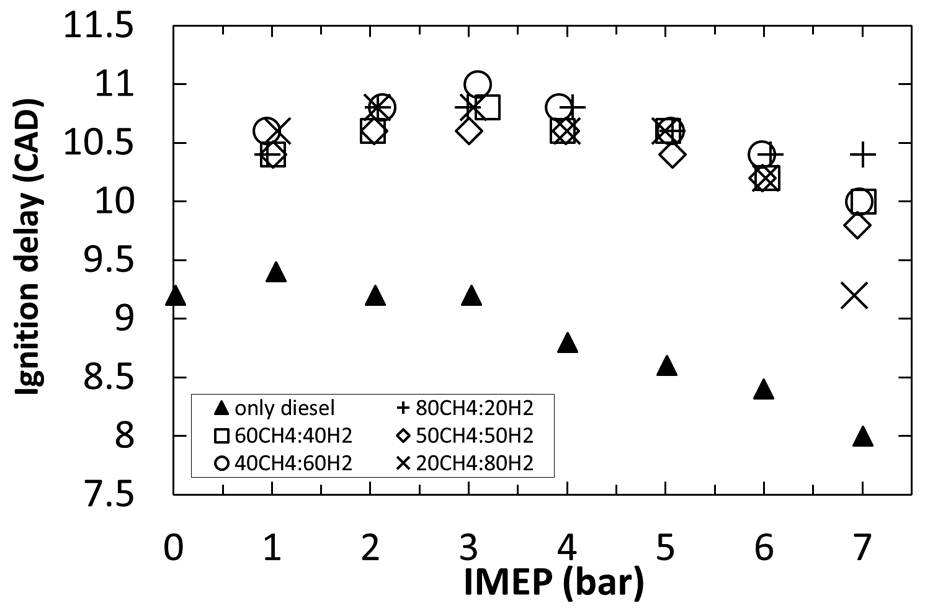
Durations of ignition delay for experiments combusting CH4–H2 mixtures pilot ignited by a fixed flow of diesel fuel for different engine loads (IMEPs) and different CH4–H2 mixture proportions.
Influences of the fuel molecular structure on engine exhaust emissions
NOx
In several studies, a strong relationship has been observed between the duration of fuel ignition delay (which is affected by fuel molecular structure as discussed in the second section of this paper) and the level of exhaust NO x emissions.4,10,31,36,37,69 Figure 9 shows the effect of the variation in ignition delay of n-alkanes and 1-alkenes on the exhaust emissions of NO x and the premixed burn fraction at constant injection timing and constant ignition timing. 31 In Figure 9(a), it can be seen that there is a linear increase in the exhaust level of NO x emissions whereas, in Figure 9(b), it is apparent that there is a concurrent increase in the extent of the premixed burn fraction with increasing ignition delay. The NO x emissions during diesel combustion are known to be strongly dependent on the in-cylinder temperature, with the thermal oxidation of nitrogen (also known as the Zeldovich 70 mechanism) dominating formation. Therefore, in Figure 9(a), the increase in NO x emissions with increasing duration of fuel ignition delay can be attributed to the increased time available for fuel–air mixing prior to the start of combustion, with a resultant increase in the premixed burn fraction (Figure 9(b)) and a higher peak heat release rate, and thus higher in-cylinder temperatures. 31
Effect of single-component fuel ignition delay on (a) the exhaust emissions of NO x and (b) the premixed burn fraction at constant injection timing and constant ignition timing.
In the co-combustion of intake-air-aspirated hydrogen with fossil diesel, some investigators have reported an increase in NO x emissions, particularly when operating with near-stoichiometric H2–air mixtures, attributed to the faster burning rates of hydrogen relative to diesel and the resulting higher in-cylinder gas temperatures.71–73 However, when operating at specific engine load conditions and for advanced diesel fuel injection timings, considerable reductions in exhaust NO x emissions with H2–diesel fuel co-combustion have been observed in other studies.74–76
Figure 10 shows the concentration of exhaust NO x emissions for H2–diesel fuel co-combustion experiments conducted by Talibi et al., 65 utilising a fixed flow rate of the pilot diesel fuel and with the engine load increased by increasing the hydrogen level. In Figure 10, for a diesel injection duration of 250 μs, levels of NO x emitted remain at approximately 20 ppm until the increase in hydrogen results in an engine load (IMEP) of 4 bar, beyond which the further addition of hydrogen results in a rapid increase in NO x emissions. A similar trend was observed in the engine tests conducted by Christodoulou and Megaritis, 77 in which up to 8 vol % of the intake air was replaced by hydrogen. No significant difference in the NO x emissions was observed at low-engine-load conditions; this was attributed to the smaller thermal boundary layer of the hydrogen flame which enhanced heat rejection and offset the higher adiabatic flame temperature of hydrogen. At higher loads, the NO x emissions were reported to increase considerably when the proportion of hydrogen energy contribution was increased, which was attributed to the elevated in-cylinder gas temperature. 77 It was suggested by Talibi et al. 65 that, for an IMEP below 4 bar, the relatively lean H2–air mixture resulted in a combustion temperature below the threshold for significant NO x formation. Only at engine loads above 4 bar was the level of hydrogen addition sufficient that the adiabatic flame temperature arising from hydrogen combustion exceeded that required for rapid NO x formation, resulting in an almost exponential rise in the NO x emissions (Figure 10). An influence of the adiabatic flame temperature on NO x emissions in the combustion of fatty acid esters, which is secondary to that of the duration of ignition delay, has been observed where the ignition delays of esters with various degrees of alkyl chain saturation have been equalised by the use of ignition-improving additives.36,44
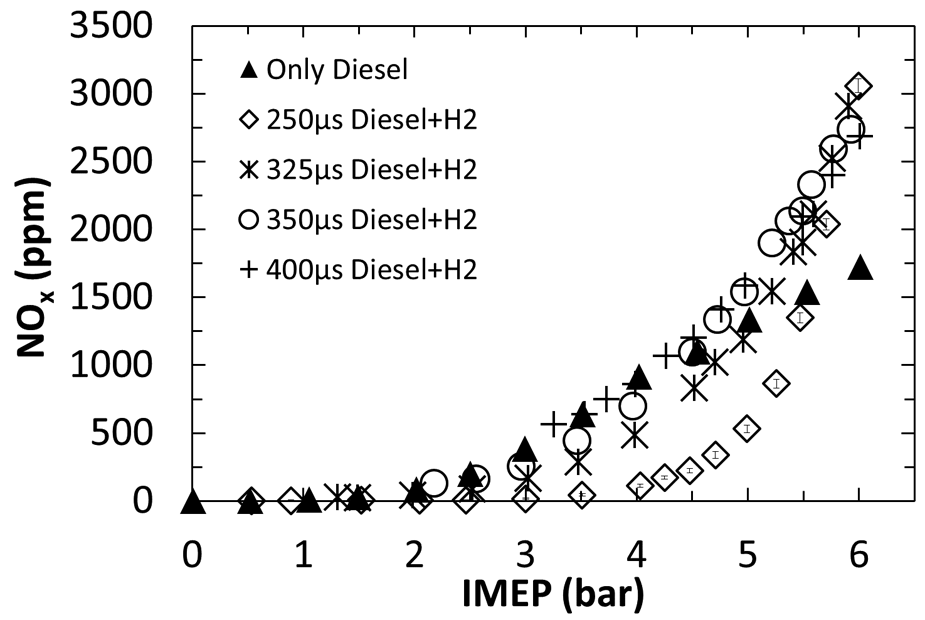
Variation in the exhaust emissions of NO x at various engine loads and various H2–diesel fuel proportions.
Similar results have been observed when co-combusting CH4–H2 mixtures with pilot diesel fuel in compression ignition engines. Increasing the proportion of hydrogen in the aspirated CH4–H2 mixture increased heat release rates during the premixed burn fraction and reduced the cycle-to-cycle variability. Furthermore, a considerable increase in emissions of NO x due to the higher adiabatic flame temperatures of hydrogen relative to methane78–80 has also been observed. Figure 11 shows the NO x emissions from a direct-injection diesel engine when combusting CH4–H2 mixtures in various proportions and using a constant diesel fuel flow of pilot ignition. 67 As observed in the case of H2–diesel fuel co-combustion, levels of NO x emitted remain at a negligible level until the stoichiometry of the aspirated CH4–H2 mixture becomes sufficient to reach the threshold temperature (1600–1800 K) which promotes rapid thermal NO x formation.
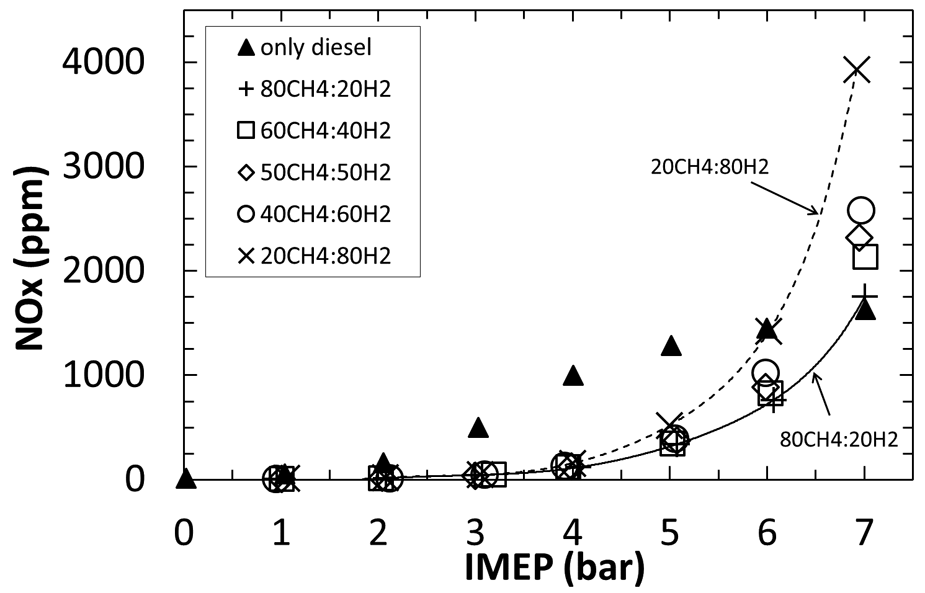
Variation in the exhaust emissions of NO x at various engine loads and various CH4–H2 mixture proportions, at a fixed flow rate of diesel fuel.
Particulate matter
While, when considering the PM emissions from compression ignition engines, an influence of fuel molecular structure via ignition delay is apparent, the relationship is not as direct as in the case of NO x formation. Observed trends of decreasing exhaust emissions of PM with increasing duration of ignition delay can possibly be attributed to a decreasing diffusion burn fraction (fuel-rich zones which account for the majority of particulate formation 68 ) and higher in-cylinder temperatures, which increase the rate of soot oxidation; 31 however, in addition to combustion phasing, the fuel composition can impact on many other parameters important to particulate formation, such as the local stoichiometry and spray characteristics. 81 In order to isolate the direct effects of fuel molecular structure from those of the combustion phasing, many experimental studies on particulate formation have been conducted under carefully controlled conditions such as laboratory flames,82–86 shock tubes87–90 and flow reactors.91–93 Smoke point tests have been widely used as a metric to determine the sooting tendencies of fuels and involve the measurement of the height at which soot is visibly seen to emerge from the tip of a wick-fed diffusion-type flame; a lower smoke point indicates a higher tendency to form soot. There is consensus that in diffusion flames the tendency of hydrocarbon molecules to form soot increases in the following order: n-alkanes < isoalkanes <alkenes <cycloalkanes < alkynes < aromatics.
Cyclic and aromatic molecules
In diffusion flame tests of the smoke point, cyclic and aromatic fuels have a far higher propensity to form soot than aliphatic fuels. 82 For example, Velásquez et al. 94 found that a diesel surrogate fuel of n-hexane, cyclohexane, benzene and toluene produced five times more soot in a diffusion flame than an n-hexane flame. The effects of the side chains of aromatic molecules are complex in that increasing the length of a side chain tends to reduce the sooting tendency, whereas increasing the number of side chains generally leads to an increase in the sooting tendency. 95
Most studies concerning the effects of fuel aromatic and cyclic compounds on diesel engine combustion have reported an increase in PM emissions with increasing content of either variety of molecule62,96–99 and, in some instances, a correlation between the engine exhaust emissions and the diffusion flame sooting tendency was observed.97–99 For example, Bryce et al. 97 and Tsurutani et al. 99 added increasing amounts of aromatic additives to a base fossil diesel fuel in tests undertaken on indirect-injection diesel engines and found that an increase in the aromatic content significantly increased the exhaust emissions of PM, with Tsurutani et al. 99 identifying that the exhaust emissions of PM increased in the following order: monoaromatics < diaromatics < triaromatics. Miyamoto et al. 96 investigated the combustion of a base fuel consisting an n-tetradecane and heptamethylnonane with the addition of various monoaromatic and diaromatic compounds. In constant-ignition-delay and constant-overall-equivalence-ratio conditions, the PM emissions increased linearly with increasing carbon-to-hydrogen ratio of the fuel, in both direct-injection and indirect-injection engines. Thus, the addition of diaromatic molecules tended to result in higher PM emissions than the addition of monoaromatic molecules because of the higher carbon-to-hydrogen ratio of the former.
However, some studies on the engine emissions of PM at constant-ignition-delay conditions have reported only a small or a negligible effect of the aromatic content of the fuel.60,100,101 Xiao et al. 62 investigated binary fuel blends of n-heptane with various levels of toluene or methylnaphthalene in an indirect-injection engine, and while the presence of both aromatic compounds resulted in a significant increase in PM emissions, in tests where toluene was gradually replaced with methylnaphthalene (and the ignition delay did not change considerably), no significant difference in the PM emissions was observed. Hellier et al. 60 combusted binary toluene–n-heptane mixtures in a compression ignition engine, where the toluene content was varied from 0% to 52% and, although an ignition-improving additive was utilised in order to equalise the duration of the ignition delay and to minimise the effects of combustion-phasing no consistent effect of increasing the toluene content on the PM emissions was observed.
Alkyl chain length and branching
Smoke point tests show that increasing the n-alkane chain length increases the propensity of the compound to form soot, as does the addition of branches to an alkyl chain. 82 Similar results have been observed in a flow reactor (at between 850 °C and 1000 °C) and shock tube studies (at near 2000 °C), where isoparaffins and cycloparaffins have been shown to produce higher yields of soot and precursor molecules (benzene and toluene) during pyrolysis. 102 However, when comparing the engine exhaust emissions of two surrogate fuels of low aromatic and sulphur content, Nakakita et al. 103 did not find an increase in PM emissions from the fuel containing 50–70% more branched structures and twice as many cycloalkanes. As the combustion phasing in the case of the engine tests of both fuels was nearly identical, it was concluded that the alkyl chain structure probably had a significant effect on the PM emissions only when the fuels had a very low aromatic content. 103
Schönborn et al. 36 investigated FAMEs with increasing alkyl chain length of the fatty acid moiety in compression ignition engine tests of single-component fatty acid esters. The engine was operated at three different injection timings (constant injection timing, constant ignition timing and constant duration of ignition delay) and, for all conditions, no discernible impact on PM emissions was found when the alkyl chain length of the FAMEs was varied between 12 carbon atoms and 18 carbon atoms (single component fully saturated FAME fuels of between C12:0 and C18:0). However, a C22 ester (C22:0) produced noticeably higher PM mass emissions, which was attributed to the higher carbon-to-oxygen ratio and higher viscosity of the fuel.
Figure 12 shows the effect of increasing the straight carbon chain length of the alcohol moiety of a saturated fatty acid ester (with a chain length of the fatty acid alkyl moiety of 18 carbon atoms) on the engine exhaust PM emissions. 40 No clear influence of substituting the methyl alcohol moiety of the fatty acid ester with the various longer-chained alcohol moieties on the duration of the fuel ignition delay was observed; 40 however, Figure 12 shows a significant increase in PM emissions with increasing chain length of the alcohol moiety in the fatty acid ester. The increase in the chain length of the alcohol moiety coincided with decreasing molecular oxygen content, increasing viscosity and increasing boiling point; 40 the changes in these properties are all expected to result in increased levels of PM formation and emissions. 81
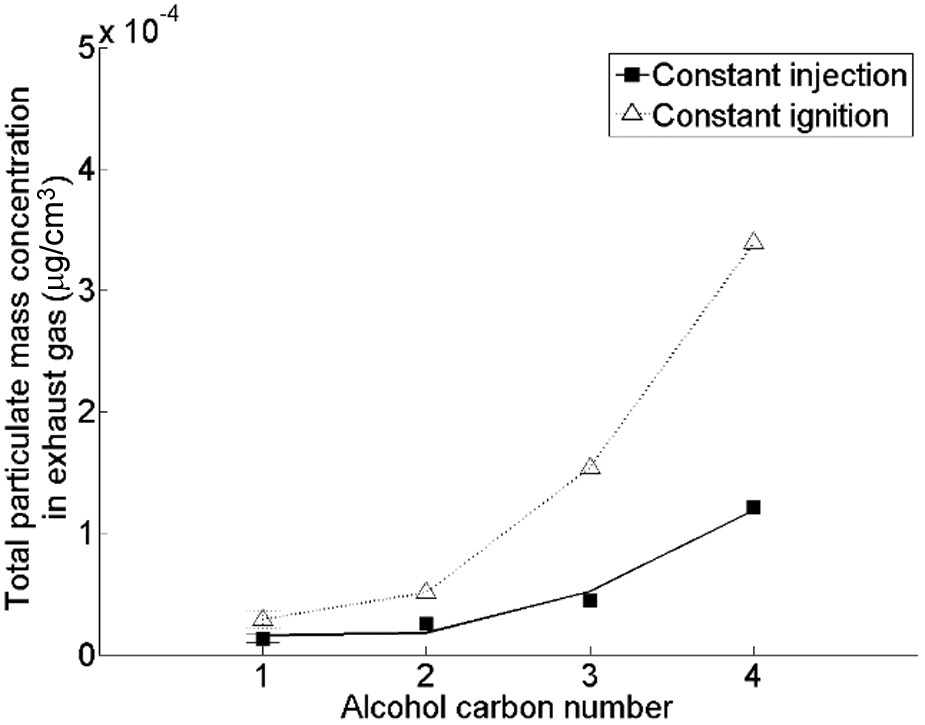
Effect of the straight carbon chain length of the alcohol moiety of the C18 fatty acid saturated ester on total particulate mass emissions.
Degrees of unsaturation and positions of the double bonds in the alkyl chain
Smoke point tests invariably show that, for molecules with comparable numbers of carbon atoms, the sooting tendency is as follows: alkanes > alkenes > alkynes. For example, Hunt 95 recorded that the fuels n-octane, 1-octene, 2-octene and 1-octyne have smoke points that are 149 mm, 99 mm, 99 mm and 24 mm respectively.
The influence of fuel molecule degree of saturation in diesel engine combustion was investigated by Schonbörn et al., 36 who varied the degree of saturation within the alkyl chain of a FAME of fixed chain length. Increasing the number of double bonds in the alkyl chain resulted in an increase in the particulate mass concentration in the engine exhaust gas, for experiments carried out at constant ignition delay timing and at constant ignition timing. However, in engine tests of binary n-octane–1-octene mixtures at constant injection timing and constant ignition timing by Hellier et al., 60 no clear influence of increasing the proportion of 1-octene was found on the particulate mass concentration measured in the exhaust.
Isotope-labelling techniques have been employed in order to establish whether double-bonded carbon atoms (C = C) tend to contribute to particulate formation to a greater extent than those that contain a single bond. Eveleigh et al. 104 carried out an experimental study where a fatty acid, namely oleic acid (C18:1), was specifically 13C labelled at the double-bonded carbon atoms (oleic acid-9,10-13C2). It was found in tube reactor and diesel engine tests that the labelled double-bonded carbon atoms tended to contribute to PM formation at approximately the same rate as the other carbon atoms in the alkyl chain of oleic acid (Table 1). Similarly, a 14 C technique employed by Buccholz et al. 105 showed that the double-bonded carbon atoms in dibutyl maleate contributed to PM formation during diesel engine combustion at the same rate as the average carbon atom in the fuel.
Movement of a single double bond within isomers of octene towards the centre of the alkyl chain was found to result in an increase in PM mass engine exhaust emissions by Hellier et al. 44 at all injection timings, and at constant ignition delay timing trans-2-octene was found to produce more nucleation-mode particles (Dp < 50 nm) than 1-octene. This observation is in agreement with the shock tube study of decene isomers by Fridlyand et al. 48 who found an increasing yield of benzene (an important precursor to particulate formation) in the speciation of reaction intermediates by gas chromatography with movement of the decene double bond from position 1 to positions 2 and 5.
Oxygen-bearing functional groups
Using the smoke point method, Barrientos et al. 86 investigated the sooting tendencies of a range of C5 oxygenated compounds. Among the mono-oxygenated compounds tested, the sooting tendencies were determined to be in the following order: aldehydes < alcohols < ketones < ethers. For dioxygenated compounds the order of sooting tendency was as follows: acids < esters < diethers.
In compression ignition engines, oxygenated fuels and fuel additives have been found to reduce exhaust emissions of PM, and the extent of the reduction is mainly dependent on the amount of oxygen present in the fuel. For example, Miyamoto et al. 106 fuelled a direct-injection compression ignition engine with diesel fuel blended with various levels of four oxygenated molecules (di-n-butyl ether, ethylhexyl acetate, ethylene glycol mono-n-butyl ether and diethylene glycol dimethyl ether). As the overall percentage of oxygen in each fuel blend was increased, a proportional reduction in the particulate mass emissions was observed and, when the oxygen content was about 25–35 % by mass of the fuel, the particulate emissions were reduced to nearly zero. It should be noted that fossil diesel fuel is typically composed of about 20% aromatics, which produce higher levels of soot than alkanes. Therefore, replacing part of the aromatic diesel fuel with a straight-chained hydrocarbon can reduce PM emissions independently of the oxygen content.
Whereas the reduction in the exhaust emissions of PM are mainly correlated with the fraction of oxygen in the fuel, there are known to be differences in the relative effectiveness of different oxygenated molecules in reducing the diesel engine emissions of PM, which have been demonstrated by a number of experimental studies52,104,106–109 and predicted by numerical kinetic modelling. 110
Isotope-labelling experiments have been carried out to aid the understanding of how oxygen influences the conversion of carbon to PM. For example, Buchholz et al. 105 found that the carbon atom directly attached to the two oxygen atoms of the ester moiety in dibutyl maleate did not contribute to PM. In further work, Mueller et al. 111 investigated, numerically and experimentally, the combustion of tri(propylene glycol) methyl ester, where oxygen is mainly present as ether linkages, and dibutyl maleate, where oxygen is bound in an ester group (Table 1). They concluded that the distribution of oxygen in tri(propylene glycol) methyl ester is more effective at reducing PM formation than the ester group in dibutyl maleate and that more than 30% of the oxygen in dibutyl maleate is unavailable for soot oxidation.
Eveleigh et al. 112 carried out a similar isotope-labelling experiment and blended a number of alcohols (ethanol, n-propanol and isopropanol) and a ketone (acetone) into binary mixtures with n-heptane to a level of 10% (molar concentration), which were combusted in a diesel engine and a flow reactor. Similar results were obtained in both systems, where in the case of the alcohols, the carbon atoms bonded to the hydroxyl groups contributed to soot formation in all cases, but to a reduced extent compared with the average carbon atom. In the case of the ketone, the carbonyl carbon atom did not contribute to particulate formation at all, indicating that the bonding and distribution of oxygen influences conversion of a molecule to particulates.
Liquid gaseous fuel co-combustion
Several investigators have reported a reduction in PM emissions during H2–diesel co-combustion. This has been attributed to a number of factors: the reduction in the overall carbon-to-hydrogen ratio of the combined H2–diesel fuel mixture, an increased oxidation rate due to a higher in-cylinder gas temperature post ignition, and the formation of OH radicals which potentially enhance oxidation of particulates.72,74,76,77,113 Figure 13 shows the percentage reduction in the total particulate mass plotted against the percentage reduction in fuel carbon supplied to the engine at constant engine loads for H2–diesel fuel co-combustion tests. 65 The proportions of diesel fuel and hydrogen were adjusted to achieve constant loads, and the percentage reductions in both particulates and fuel carbon were calculated against the values obtained with the engine operating on only diesel fuel (no hydrogen addition). It can be seen in Figure 13 that, for an engine IMEP below 4 bar, the reduction in PM emissions lies in the top half of the graph, which implies a reduction in the PM emissions beyond that obtained by simple fuel carbon displacement. This suggests that, at engine loads (IMEP) below 4 bar, an increase in the H2–air stoichiometry (or decrease in the diesel fuel) promotes soot oxidation (or reduces initial soot production), and this offsets any increase in soot formation due to reduced intake oxygen levels. At 6 bar IMEP, the reductions in the PM emissions lie in the bottom half of the graph, suggesting that, although substitution of hydrogen results in a small decrease in particulates, this reduction is considerably lower than expected for the drop in fuel carbon supplied to the engine (Figure 13). Furthermore, at similar high hydrogen substitution levels, several other studies have reported an increase in PM emissions.72,114 These observations have been attributed to the displacement of significant amounts of intake air (thereby reducing the in-cylinder oxygen concentration), which causes a reduction in rates of fuel oxidation, hence resulting in an increase in PM emissions.65,72,114
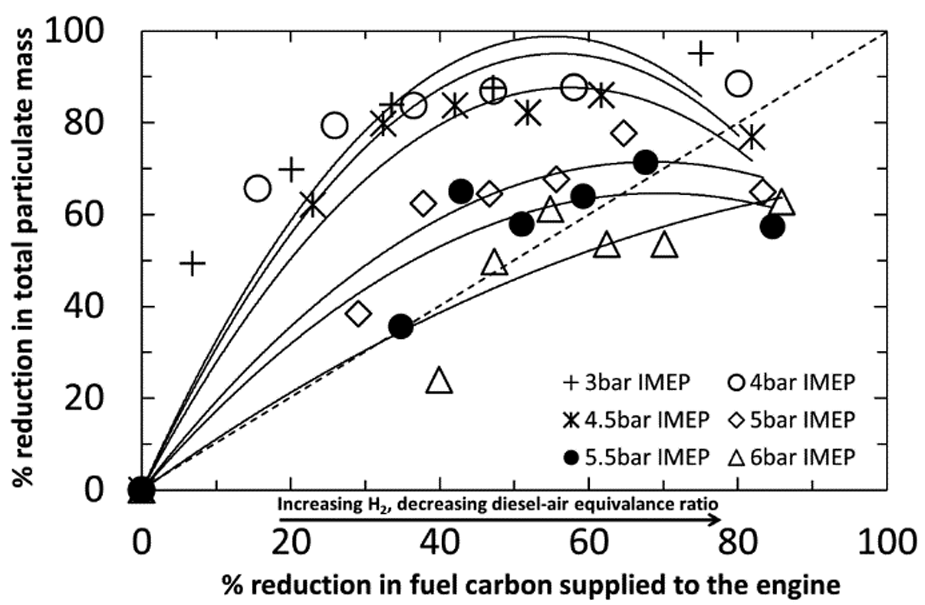
Comparison of the percentage reduction in total particulate mass with percentage reduction in the carbon content of the combustible mixture at constant engine loads for H2–diesel fuel co-combustion experiments.
Conclusions
From compression ignition engine tests on single-component fuels, binary fuel mixtures and pilot-ignited H2–CH4 methane combustion, the following conclusions regarding the impacts of fuel molecular structure on diesel engine combustion and emissions characteristics can be made:
Increasing the length of a straight alkyl chain decreases the duration of ignition delay by enhancing the rates of hydrogen abstraction and isomerisation during low-temperature reactions. This is apparent in a variety of species that contain alkyl moieties, including n-alkanes, alkenes, fatty acid esters and alcohols.
Introducing double bonds into an alkyl chain increases the duration of ignition delay by reducing the rates of low-temperature branching reactions. Moving a double bond towards the centre of a fuel molecule can result in both a decrease and an increase in the duration of ignition delay, dependent on the overall alkyl chain length and the presence of oxygenated functional groups.
Addition of oxygen atoms to the functional group of a fuel molecule increases the duration of ignition delay. However, the introduction of oxygen atoms within an alkyl chain as ether linkages, or the addition of an oxygenated functional group where the alkyl moiety is of poor ignition quality, can decrease the duration of ignition delay.
The presence of aromatic molecules in binary fuel mixtures significantly increases the duration of ignition delay by inhibiting rates of alkyl chain low-temperature reactions. In mixtures of fossil diesel combustion with intake aspirated hydrogen or CH4–H2, increasing the levels of the gaseous fuel affect the fossil diesel ignition only by displacement of the intake oxygen.
The duration of ignition delay is the primary influence of fuel molecular structure on the exhaust emissions of NO x . Increased duration of ignition delay results in higher levels of thermal NO x production via a larger premixed burn fraction and subsequently higher in-cylinder temperatures.
The emissions of NO x from H2–fossil diesel co-combustion are thermally driven, in that NO x production from hydrogen combustion commences only at a stoichiometry sufficiently rich for resultant combustion temperatures to exceed those required for significant NO x formation.
When the combustion phasing is held constant, increasing the number of aromatic rings within a hydrocarbon molecule increases PM emissions, as does increasing the alkyl chain length where such changes result in increases in the physical properties (such as the boiling point and the viscosity) which impede fuel–air mixing.
Hydrogen promotes PM burnout because of elevated temperatures and higher oxidation rates at low engine loads; however, at higher loads, where the hydrogen addition displaces intake oxygen, PM increases as lower oxygen availability increases particulate formation and reduces rates of PM oxidation.
It can be seen that relatively minor changes to the molecular structure of fuels for compression ignition engines can significantly influence the combustion and emissions characteristics. Therefore, the current and ongoing study of the production routes of alternative fuels is an opportunity to utilise this understanding as to the effects of fuel molecular structure to influence the development of these processes so that the resultant fuels are optimised for clean and efficient operation of compression ignition engines. Furthermore, such understanding can also be exploited for the production of designer fuels for future low-pollutant combustion concepts, 115 e.g. those that require reduced durations of ignition delay or variable control of the ignition timing.116,117
Footnotes
Appendix 1
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the UK Engineering and Physical Science Research Council (grant numbers EP/M007960/1 and EP/M009424/1).