
Abstract
Keywords
All pharmaceutical products are continuously experimental, observed and tracked by pharmacovigilance systems worldwide.
1
Introduction
Strong science, characterised by open mindedness, objectivity, curiosity and freedom of debate, can be corrupted by capitalist opportunism, deception, political ideology and censorship. Regulatory protections are required for good science to flourish. Corporate enthusiasm and authoritarian policy directives, such as vaccine mandates, must be balanced with humane medical ethics and protection of individual autonomy.
The global pharmaceutical industry has grown in recent decades and now represents one of the most valuable in the world. Revenue of the worldwide market in just 2 decades has risen from 390 billion USD (2001) to 1482 billion USD (2022). 2
New additions to the global marketplace appear with entrepreneurial enthusiasm. Yet withdrawals of these products are also significant. In the last 7 decades, from 1953 onwards, more than 462 medicinal products have had to be recalled from sale because of adverse drug effects that frequently include fatalities. The median interval between the first reported adverse reaction and the year of first withdrawal for a drug is 6 years (IQR, 1–15). 3
Globally, whether drugs are recalled or not, pharmaceutical industry violations have become a multibillion-dollar industry of litigation, legal fees, and court penalties. Some of the most impressive corporate criminal trials include
4
: • • • • •
Direct to public commercials in the USA for legal support are now widespread, e.g.
4
: “Call a Dangerous Drug Attorney at O’Connor, Acciani & Levy. If you believe you were harmed after using a certain pharmaceutical product, call a skilled dangerous drug attorney for help in starting a personal injury claim.”
In this selective narrative review, our goal is to consider some of the milestones in drug recall from the market, litigation for, and republication of, hidden data, and potential lessons that may be learnt. We assess recall of various pharmaceutical agents, proven over time to be monumental events. In particular, we focus on the cases of Merck’s Vioxx (rofecoxib), and the new gene-based COVID-19 vaccines.
Results
Diethylstilbestrol (DES)
Marketed widely in the 1950s and 1960s, diethylstilbestrol (DES) (Eli Lilly), prescribed by the medical profession for prevention of miscarriage, led to extensive harm that would prove fatal for some and would span generations. Supplied to millions of pregnant women over 3 decades, DES became the first identified cause of “prenatal drug-induced cancer in humans”. The drug was recalled in 1971. The full intergenerational impact of these prescriptions is still not known. 5
Thalidomide
Thalidomide is one of the saddest chapters in pharmaceutical history and an example of how premature safety claims can have tragic consequences. Created as a sedative and marketed in Germany in 1957 by Chemie Grünenthal, thalidomide would soon be launched in the UK (Distillers, UK), and many other countries would follow. At this stage, the first thalidomide-affected baby had already been born in Germany, 25 December 1956, to a Chemie Grünenthal employee. By 1958, thalidomide was licensed and promoted in the UK as a “wonder drug” to treat headaches, insomnia, and nausea in pregnant women – advertisements emphasised safety, with catch phrases such as “non-toxic” and “no known toxicity”.
The first publication to link thalidomide and birth defects appeared in 1961 in The Lancet, as a letter from an Australian, William McBride. 6 This same year the drug was formally withdrawn in Germany and in the UK, the Thalidomide Society was established in the UK, and efforts began to secure compensation for victims. In 1968, Chemie Grünenthal was brought to trial in Germany, charged with intent to commit bodily harm and involuntary manslaughter, but in 1970 this trial was ended prematurely by the German government, who stated that it was “not in the public interest”. 7
Efforts have continued in the UK to secure compensation from the 1970s through to the present. It was only on 29 November 2023 that the Australian Prime Minister announced a “formal national apology to all Australians impacted by the Thalidomide Tragedy”, more than half a century on from the earliest harms. 8
Through the diligent work of FDA scientist Frances Kelsey, who demanded further safety trials prior to market authorisation, thalidomide was never approved for release in the USA. She protected an entire nation. 9
Paroxetine
The Selective Serotonin Reuptake Inhibitor (SSRI) antidepressant, paroxetine, became a very successful commercial product for SmithKlineBeecham (SKB) (later GlaxoSmithKline, GSK). In the late 1990s, the company conducted two randomised, controlled trials in adolescents with depression (Study 329 & Study 377). Company documents, subpoenaed through litigation, reported that Study 377 “failed to demonstrate any separation from placebo” and consequently the company had “no plans to publish data from Study 377”. Study 329 showed “trends in efficacy” but the differentiation from placebo “was not statistically significant”. 10 This Study 329 was ghost written and then published by Keller and 21 co-authors in 2001, with the conclusion that paroxetine was “generally well tolerated and effective” for adolescents with depression. 11 Although SKB/GSK decided not to present the studies’ data to the FDA for a label change to treat adolescent depression, they used the Keller et al. publication to promote off-label prescriptions for depressed teens. Later, independent researchers gained access to raw data from Study 329 and found increased suicidality and no significant efficacy. 12 Despite calls for retraction of the original Study 329 publication, the Journal of the American Academy of Child and Adolescent Psychiatry (JAACAP) has refused to do so. 13
GSK suppressed negative data about their drug paroxetine and effects on depression and suicide. An internal GSK document advised staff to withhold data that indicated paroxetine had no beneficial effect in adolescents. 14 In 2012, GSK pleaded guilty to fraud allegations and failure to report safety data, with payment of $3 billion in criminal fines, the largest fraud settlement in US history at the time. 15
There have been further disputes over the increased suicidality caused by SSRIs in adolescents and young adults, with calls to remove the FDA Black Box label. However, both Study 329 data re-analysis 13 and separate further data support continuation. 16
This GSK paroxetine chapter is by no means an isolated case of hidden data. In 2015, Eli-Lilly and Takeda Pharmaceuticals were fined $2.4 billion USD for concealment of the carcinogenic effects of pioglitazone (Actos). 17
Avandia (rosiglitazone)
Avandia (rosiglitazone) gained FDA approval for management of diabetes in May 1999 and was widely prescribed for control of blood glucose, until it was shown to increase risk of myocardial infarction by 43% and increase risk of death from cardiovascular causes by 64%. 18 In May 2007, Steven Nissen of the Cleveland Clinic published controlled trial data that showed, in the rosiglitazone group, as compared with control, the odds ratio for myocardial infarction was 1.43 (95% confidence interval (CI), 1.03 to 1.98; p = 0.03), and the odds ratio for death from cardiovascular causes was 1.64 (95% CI, 0.98 to 2.74; p = 0.06). 19
In July 2007, a panel of FDA advisers voted 22 to 1 against removal of Avandia from the marketplace. As late as 2009, GSK continued with promotion of Avandia as “safe and free from cardiovascular side effects”. 20 In contrast, by February 2010, a US senate finance committee was able to conclude that GSK had “full knowledge of the cardiac risks of Avandia in late 2004 or early 2005”. David Graham, FDA scientist, has estimated combined US heart attacks, strokes and deaths caused by Avandia to be in the order of 100,000 events. 21 The drug was removed from the European market in September 2010, based on cardiovascular risks, and remains banned to this day.
Pursuit of surrogate end points can be dangerous, exemplified here with a focal target of blood glucose control, yet accompanied by significant adverse events.
While such corporate products and medical prescriptions as diethylstilbestrol, thalidomide, paroxetine and rosiglitazone are now infamous chapters in medical history, still greater events loom over more recent history, and we consider two of these, Merck’s Vioxx (rofecoxib) scandal, and the roll out of gene-based COVID-19 vaccines.
Vioxx (rofecoxib)
Developed by Merck, the cyclooxygenase-2 (COX 2) inhibitor Vioxx (rofecoxib) marketed as a non-steroidal anti-inflammatory drug (NSAID) for pain relief in 1999, obtained FDA approval (21 May 1999) based on equivalence to other NSAIDs in short term use. Efforts to explore long term value in rheumatoid arthritis further supported sales, with fewer gastrointestinal side effects when compared with typical NSAID naproxen. 22
In this VIGOR paper, 22 Merck concealed adverse cardiovascular events in the Vioxx arm of the study that would prove to be a serious statistical signal. Just prior to publication, Merck informed the FDA of three adverse cardiovascular events, published on an FDA website, but The New England Journal of Medicine (NEJM) article was neither retracted nor corrected.
The full VIGOR data unmasked high rates of cardiovascular events with Vioxx (rofecoxib) compared to naproxen, with a relative risk of 2.38 (95% CI 1.39–4.00) for rofecoxib against naproxen over a 12-month study period. 23 The time lag between initial FDA approval and the appearance of this more complete VIGOR trial data in print was over 18 months.
Initial responses to this data from Merck included claims that naproxen had a protective effect against heart attacks and strokes, that was not possessed by Vioxx, and that the increased cardiovascular risks seen with Vioxx occurred only in people with known cardiovascular disease. 24 This was later found to be untrue, once data for healthy individuals who had suffered harm on Vioxx had been uncovered.
Merck tried to influence lead American physicians with support and finance for research, and they defamed, withdrew support, and tried to discredit or “neutralise” those who failed to promote use of Vioxx, a matter uncovered by the Federal Court in Melbourne, Australia. 25 In contrast, the Chair of the Study Data and Safety Board (SDSB) for the study, Michael Weinblatt, owned $72,000 in Merck stock and was on a $60,000 contract for 12 days’ work for the company. 26
Internal Merck emails are now known to have shown as early as 18 November 1999 (unblinded minutes), that an interim safety analysis of VIGOR showed excess deaths and cardiovascular adverse experiences – 79 cardiac events for rofecoxib compared with 41 for the control group on a traditional NSAID, naproxen.26,27 Yet Merck made a press release on 22 May 2001, entitled “Merck Reconfirms Favourable Cardiovascular Safety of Vioxx”. Merck even created a “fake journal” with the medical publisher Elsevier: The Australasian Journal of Bone and Joint Medicine, with six issues between 2002 and 2005, that collated articles favourable to Merck’s drugs Vioxx and Fosamax. 28
The FDA appears to have been complicit with Merck in early suppression of the adverse event data of VIGOR. Eventually the FDA did instruct Merck (April 11th, 2002) to include a precaution about cardiovascular risks in their package insert. 24 Dr David Graham, an FDA scientist in its Office of Drug Safety, revealed this interplay in his testimony to the US Senate (below).
Vioxx remained on the market until the completion of the APPROVE study in 2004. The intention was to promote use of Vioxx to treat polyps of the colon. But again, the drug demonstrated at least double the cardiovascular risk compared with placebo, this time in a patient population considered to be at low risk of cardiovascular disease. 29
Merck announced withdrawal of Vioxx on 30 September 2004, the largest prescription drug recall in history to date.
Over 20 million people in the US are believed to have taken the drug, of whom an estimated 88,000 to 139,000 suffered myocardial infarctions, with 30–40% fatality rate (testimony of Dr Graham to the US Senate). 30 His figures on estimated cardiac arrests were also published in The Lancet, despite opposition from the FDA. 31 Dr Graham further testified to the Senate that conflicts of interest at the FDA had delayed the Vioxx recall. 32 Discovery documents in litigation reveal corporate pharma may conceal data early, at any cost to achieve market growth.33,34 Here the FDA appeared complicit and slow to withdraw the product.24,35 Published in the NEJM, prominent cardiologist Eric Topol included strokes as well as myocardial infarctions to estimate 160,000 events per 10 million people prescribed Vioxx, and he noted a global cohort of up to 80 million had been prescribed Vioxx. 24
By August 2005, 13,000 class action lawsuits had been filed against Merck. By November 2007, Merck had created a settlement fund of $4.85 billion USD, the largest ever in US history at the time. Merck agreed to compensate victims in exchange for a no-fault agreement – specifically, no legal admission of fault. Yet payment of $4.85 billion USD in compensation to claimants could clearly be interpreted as an admission of fault.25,26
When the Vioxx scandal broke, Merck had a capital market value of between $40 and $50 billion USD. Despite the greatest drug scandal in the world, enormous fines and atrocious damage to image, Merck has continued to grow in the last 2 decades and has increased its value six-fold to over $300 billion USD.
COVID-19 gene-based vaccines
Initially marketed December 2020, as Emergency Use Authorisation (EUA) in the USA, and provisional authorisation in Australia and other nations, the gene-based COVID-19 vaccines of modified mRNA type, (Pfizer-BioNTech’s BNT162b2, Moderna’s mRNA-1273) and viral-vector-DNA type (AstraZeneca’s ChAdOx1-S, Janssen’s Ad26.COV2.S, Gamaleya’s Sputnik V) have constituted the majority of over 13 billion doses of all COVID-19 vaccines.36–41 In contrast, COVID-19 vaccines that employ traditional well-tested inactivated virus or recombinant protein antigen-based technologies have been utilised mainly in a few non-Western nations (e.g., Bharat Biotech’s Covaxin, Sinovac’s CoronaVac, Cinnagen-Vaxine’s SpikoGen, Cuba’s Genetic Engineering and Biotechnology Centre’s Abdala). 42
Purposed for protection against transmission of the SARS-CoV-2 virus and reduced disease severity, official sales narratives included – “safe and effective”, and “millions of lives saved”. Indications of serious harm appeared from 2021 with record high adverse event reports to pharmacovigilance. These included suspected death reports as indicated by VAERS data
43
(Figure 1), peer-reviewed VAERS and EudraVigilance data,
44
excess mortality above expected from collation of official death statistics by Our World in Data
45
and insurance data for excess mortality and disability
46
correlated with COVID-19 vaccination. Montano (2022) compared COVID-19 vaccines (Janssen, Moderna, Pfizer-BioNTech) with influenza vaccines, and found extremely high elevated relative risk for serious and fatal adverse events across most organ systems [
44
, in Table 3b]. Excess mortality is defined as mortality above normal background rates at ourworldindata.org which is under the jurisdiction of Oxford University, UK. Reported suspected deaths from vaccines to VAERS since 1990 comparing all other vaccines combined with COVID-19 vaccines. From VAERS Analysis
43
(with permission).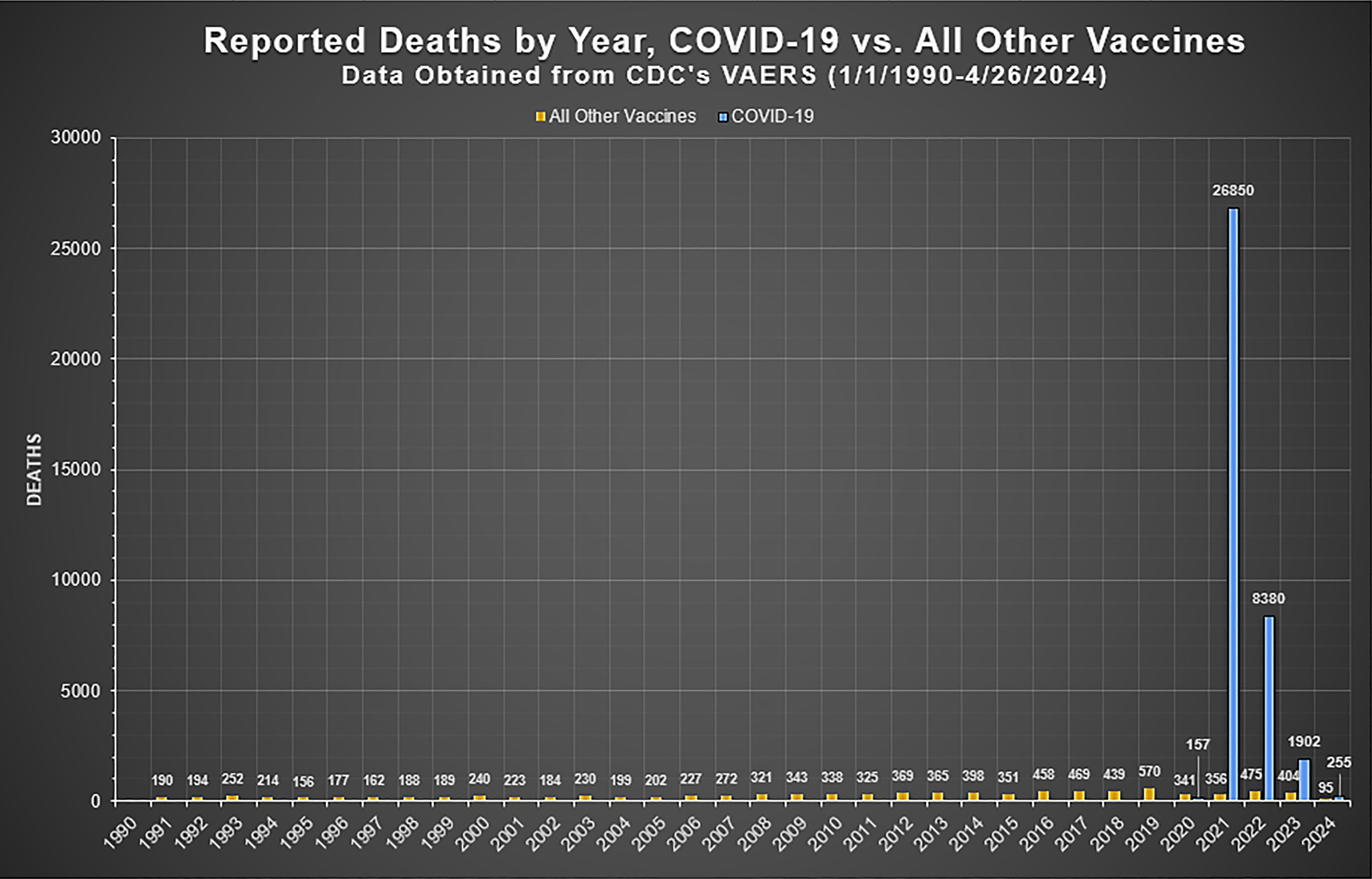
Market restrictions on recommendations began September 2022, with COVID-19 booster vaccines generally limited to over age 50 and the vulnerable in Nordic nations and Switzerland, e.g., the Danish Health Authority declared it was “no longer possible … for children and adolescents aged under 18” to get the COVID-19 vaccine “from 1 September 2022”.
47
By contrast, the USA, Canada, Australia and some other nations still market for children. The key failure is to have mandated injections in young and healthy adults; these mandates correlate with excess mortality.44–46 A recent peer-reviewed study in BMJ Public Health on excess mortality from 47 Western nations, finds over three million excess deaths from January 2020 to December 2022. Notably, when stratified by year, the highest number of excess deaths was reported in 2021, the year in which mass vaccination began. Especially in late 2021 which saw imposition of vaccine mandates in many nations (first graph p. 5).
45
Additional lessons potentially are that rushed “warp speed” development of novel technologies is unwise; narrative and groupthink can distort judgement; suppression of clinical trial data is harmful; heightened active pharmacovigilance must be encouraged.48–50 COVID-19 virus vector DNA and mRNA vaccines: mechanism of action. From Pichichero ME (with permission).
51
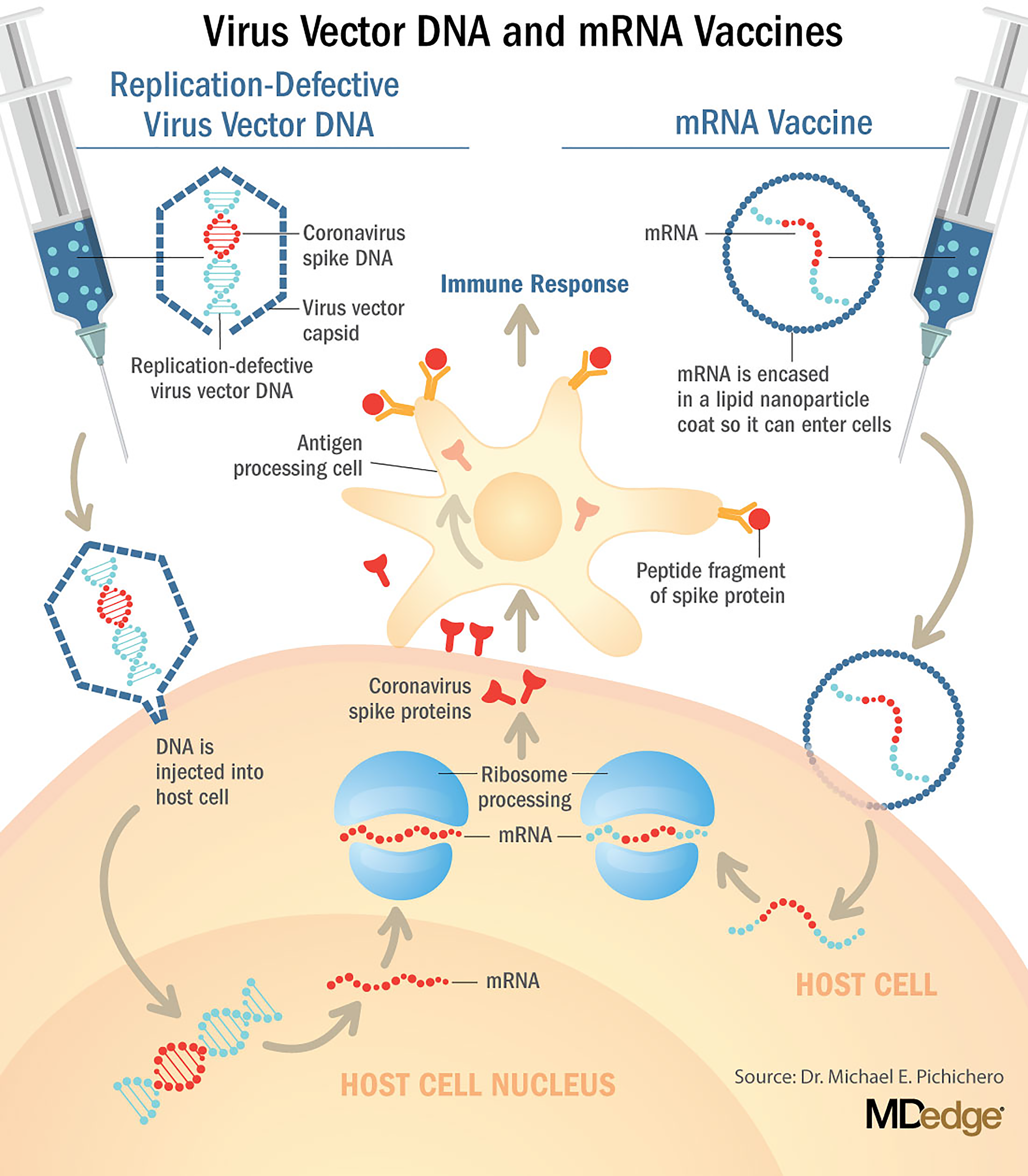
Use of the term “vaccine” for novel experimental agents that deploy gene codes may convey a false sense of assurance in the absence of supportive data and thus may mislead. In pharmacological design terms, these products are “pro-drugs”. 51 They must enter cells and undergo translation of genetic code before intended outcomes unfold 52 (Figure 2), and in this sense they operate as “synthetic viruses”. 53 Unintended consequences are thus possible.53–57
A systematic review of the peer-reviewed literature: “Serious harms of the COVID-19 vaccines: a systematic review” by Gotzsche and Demasi (2024) [
58
preprint] found that with the notable exception of Fraiman et al.,
59
“most studies were of poor quality” (abstract) and used methodologies such that “serious harms are vastly underreported” (p. 7). They conclude: Adenovirus vector vaccines increased the risk of venous thrombosis and thrombocytopenia, and the mRNA-based vaccines increased the risk of myocarditis, … serious neurological harms (occurred), which are likely due to autoimmune reaction. … Severe harms were underreported in the randomised trials [published in the NEJM].
As authorisation and promotion of the COVID-19 mRNA vaccines continue, the authors call for randomised trials of COVID-19 booster doses in high-risk groups that thoroughly examine serious adverse events. 59 The authors also state that “Authorities … do not consider that the balance between benefits and harms becomes negative in low-risk groups such as children [and those with natural immunity]” (abstract). This point has been well made by Bardosh et al. (2024) 60 who argued against universal vaccine mandates and noted that based on the Pfizer-BioNTech vaccine booster trial data, 61 to prevent one COVID-19 hospitalisation, 18.5 students would suffer a serious adverse event. 60
These products are novel and experimental, whether modified mRNA gene codes encased in lipid nanoparticles (LNP) (Pfizer-BioNTech and Moderna), or viral-vector-DNA gene codes encased in adenovirus shells (AstraZeneca, Janssen, Sputnik V). These gene sequences produce the spike protein antigen of the SARS-CoV-2 virus, which must be extruded from the cell surface as foreign protein to stimulate an immune response. This is a new mechanism for public vaccination, completely distinct from traditional vaccine technologies.
Moreover, rigorous assessment of long-term safety of these experimental gene-based products has been effectively sabotaged by the early dissolution of the placebo arm in phase III clinical trials. 62 Despite this, the interim and extensive publication of these abbreviated clinical trials in the NEJM has been used to support marketing and the public health message of “safe and effective”.
In terms of efficacy, failure to prevent infection or transmission of the COVID-19 variants63–66 eventually led the US Centers for Disease Control and Prevention (CDC) to reinvent their definition for “vaccine” as no longer the provision of “immunity,” but as “protection” against disease severity67,68 – now a narrative challenged by more recent data. Promotion of the belief that millions of lives would be saved by these agents has been based on hypothetical, predictive epidemiological models which have a track record of miscalculation.46,53,73 Official data from New South Wales state in Australia by late 2022 during the omicron variant wave did not concord with the message that these agents prevent serious disease or death, and even suggested the opposite. 53
For the wealthy western nations who have utilised these novel agents in particular, the haste and scale of development, production, distribution, and administration is unprecedented. 69 Yet haste, especially at “warp speed”, should be alien to good medical science. It is likely that novel technology, haste in vaccine development and mass production all contributed to the reported phenomenon of “batch toxicity” based on official pharmacovigilance data. 70
Key failures – Coercion and mandates, ridicule of educated hesitancy
Perhaps the greatest failure of gene-based vaccine use is the political act to mandate therapy. Mandates are relatively rare in medical history. Vaccine passports to engage in normal life resemble measures under totalitarian rule. The deadlines for COVID-19 vaccine mandate compliance correlated closely with excess morbidity and mortality.1,44,46
Given the novel nature of gene-based COVID-19 vaccines, it may be no surprise that “vaccine hesitancy” among those with tertiary qualifications was highest with PhD doctorates (January–April 2021, 14.6%), 71 and among healthcare workers was highest for “emergency medical technicians/paramedics” (April–May 2021, 45.4%). 72 Reflective of both research and coalface clinical experience. This could thus be referred to as “educated hesitancy”, found in a cohort most familiar with the imperfections of corporate sponsorship, market authorisation and medical literature, and a cohort on the frontline. Educated hesitancy towards these products has been ridiculed. It is particularly tragic that mandates have been applied to the young, fit, and healthy in our workforce, at minimal risk from the coronavirus itself, some of whom have paid the ultimate price with loss of life.43–46 In fact, at a global level the median pre-vaccination infection fatality rate (IFR) was estimated at 0.03% for the 0 – 59-year-old population, while for children aged 0–19 years the median IFR was 0.0003%. 73 These observations indicate that children and adolescents are essentially at zero risk of COVID-19 mortality.
The limitations in the peer-reviewed literature to identify and quantify the harms of the gene-based COVID-19 vaccines [58, preprint], means greater consideration must be given to analyses of public datasets of passive and active pharmacovigilance and insurance and actuarial data. A graph of Western Australian Vaccine Safety Surveillance (WAVSS) (Figure 3 in our prior paper) 1 illustrates this, and it should be noted that due to remote geography and border closures, the state of Western Australia was essentially free of the SARS-CoV-2 virus in 2021. 1
Similarly, a strong temporal correlation was evident between the imposition of COVID-19 vaccine mandates for employment in the third quarter of 2021 in the USA and high excess mortality for working age (25–64 years old) Americans, in the data collated by the US Society of Actuaries Research Institute, as shown in the table from Cause Unknown by Edward Dowd
46
(p. 80) (Figure 3). Table 5.7 Excess mortality by detailed age band. From p.80 Dowd E (2022)
46
(with permission).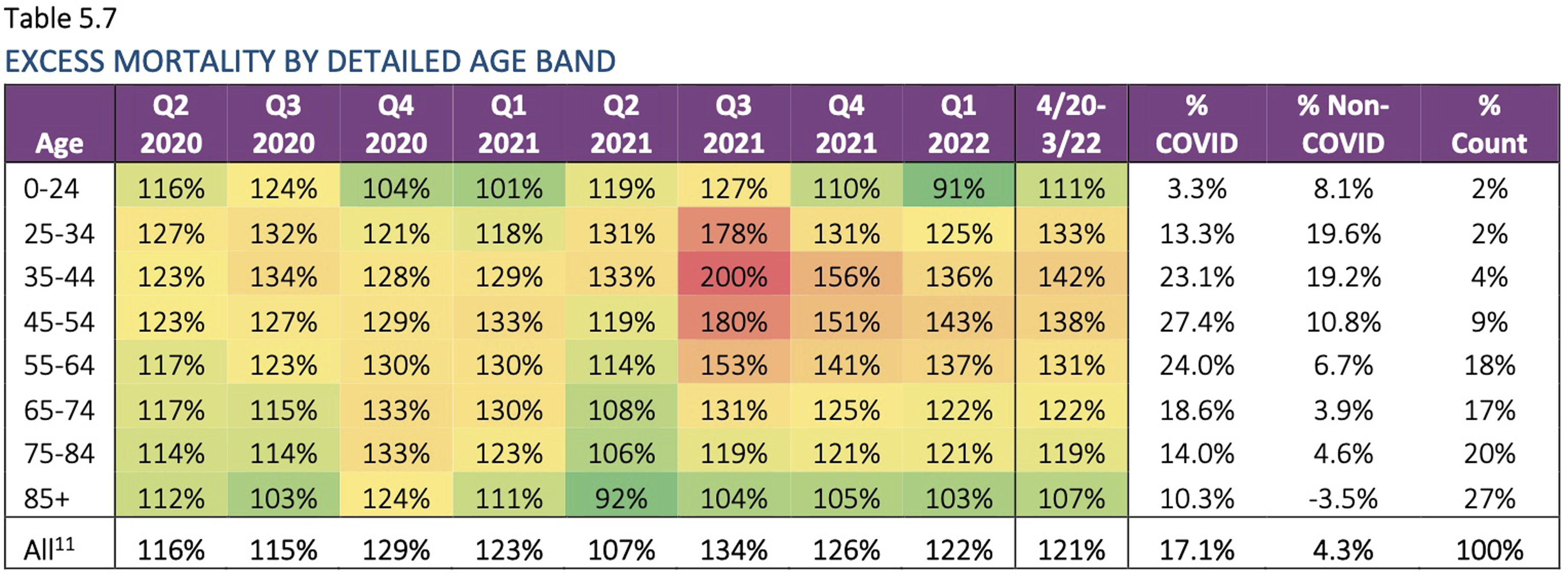
With Vioxx, the key publication of the VIGOR clinical trial in the NEJM excluded three subjects with severe cardiovascular adverse events, a data suppression that obscured the true risk. Similarly with the phase III clinical trials for the Pfizer, AstraZeneca and Moderna COVID-19 vaccines it is now known that three subjects with serious adverse events were excluded [49,58 preprint, 74 ] from key papers36,37,39 in the NEJM, which influenced health policy globally. These omissions occurred in the context of a non-random excess of 251 exclusions from the vaccine arm compared to placebo arm (311 vs 60) in the Pfizer clinical trial 75 and reported unblinding at one of the clinical trial sites. 76
Two phase III clinical trials subjects who suffered severe adverse events from the vaccine arms of the Pfizer-BioNTech trial and the AstraZeneca trial [49,58 preprint], and one from the Moderna trial 74 came forward to say their adverse event data was not published in the NEJM peer-reviewed papers of the clinical trials, and likely not reported to the FDA either. In the case of AstraZeneca, this was despite appeals to the journal. 49 A further case of a 12-year-old in the adolescent Pfizer COVID-19 clinical trial, suffered permanent severe polyneuropathy and is wheelchair bound [ 58 preprint,77,78], is recorded in the NEJM paper as “functional abdominal pain”.
Additionally, the Pfizer-BioNTech phase III trial report submitted to the FDA for Emergency Use Authorisation listed 2 deaths in the mRNA vaccine arm and 4 deaths in the placebo arm. However, documents released under court order revealed a further 4 deaths in the vaccine arm and 1 death in the placebo arm, to give the total number of deaths before the data cut-off date actually 11 (6 vaccine, 5 placebo) versus the 6 disclosed. Closer examination of relevant documentation available for each patient showed a pattern of delay in death notification, a clear violation of trial protocols and legal requirements. 48 By the end of the truncated Pfizer phase III trial there were 21 deaths in the vaccine arm and 17 in the placebo arm and the difference was accounted for by cardiovascular mortality.
Discussion
In this selective narrative review, we have chosen some of the most well-known drug recalls and data suppression scandals. We have sought insights from these events that may help better appraise the current gene-based COVID-19 vaccines, which have together formed the largest ever launch of novel pharmaceutical product in history.
Medical research
Quality of research in medical science is problematic. The scientific “replication crisis”, which is also a publication crisis, has been studied, debated and recognised in surveys of scientists79,80 ever since Ioannidis’ highly cited 2005 paper asserted that at least half the published medical literature may simply be wrong. 81 The crisis rests on pressure to publish, failure to publish negative and/or unfavourable data, lack of data transparency, poor methodological design of studies, statistical errors, carelessness, inexperience of peer reviewers and editors, commercial interest, ideological biases, failure to declare conflicts of interest and fraud.81,82 Tanver et al. noted lack of data transparency in the COVID-19 vaccine trials 83 and cast doubt on their use in public health, as did senior and chief-editors of the BMJ. 50
Distorted data, particularly due to commercial bias, is regularly published in medical journals. A Cochrane Review meta-analysis found odds ratios exist for a sponsored drug trial to find results, (OR 2.05) and provide conclusions (OR 2.69) in favour of the drug versus an independent trial for the same agent. 84
Corporate integrity and data transparency
Concerns exist related to data transparency, access to raw data, and the potential for hidden data, deleted data or indeed failure to record data.10,12,15,24,30,33,34,49,50,74–90 The track record of the pharmaceutical industry in these areas has been weak. Internal industry documents released after criminal convictions of the companies concerned, reveal a systemic pattern geared towards “marketing-based medicine” that is at odds with “evidence-based medicine”. 33
Among many examples, an internal AstraZeneca email discussed “burial” of data from four clinical trials. We quote John J A Tumas, Publications Manager, AstraZeneca, 6 December 1999, There is pressure from outside the industry to provide access to all data from clinical trials conducted by the industry; thus far we have buried trials 15, 31, 56 and are now considering COSTAR.
90
Illusion of evidence-based medicine
Jureidini and McHenry, in a prominent article in the BMJ asserted that Medicine has been “corrupted by corporate interests, failed regulation and commercialisation of academia”, to cause an “illusion of evidence-based medicine”. 85 The evidence base for clinical and public health decisions has long been corrupted, in the view of former chief-editors of The Lancet, 91 the BMJ 86 and NEJM. 87 Peer review cannot possibly police commercial and ideological conflicts of interest.
Pharmaceutical companies, publication and statistics
Manipulation of statistics in the medical literature has been lamented. 18 Widespread promotion of relative rather than absolute risk and use of surrogate endpoints are examples.18,75
Concerns exist over the transparency of COVID-19 mRNA vaccine trial data. Available figures from Pfizer and Moderna trials listed at clinicaltrials.gov have been evaluated (NCT04368728 and NCT04470427). As originally published in NEJM, the Pfizer and Moderna mRNA COVID-19 vaccine interim phase III clinical trial reports suggested a favourable risk/benefit ratio. Based on exactly the same data, Fraiman and colleagues publish in Vaccine that: mRNA COVID-19 vaccines were associated with an excess risk of serious adverse events of special interest of 10.1 and 15.1 per 10,000 vaccinated over placebo baselines of 17.6 and 42.2 (95% CI −0.4 to 20.6 and −3.6–33.8), respectively.
From which they conclude a need for formal risk-benefit analyses. 59
The FDA has been publicly criticised for their slow response to follow up potential increases in serious adverse events in elderly people related to Pfizer’s mRNA COVID-19 vaccine [ 58 preprint,92,93].
There are even indications that initial clinical trial work, published in the NEJM, may have been performed with mRNA products that differed from those eventually mass-produced. The clinical trial mRNA gene codes were created by PCR “Process 1” technology, but the vials for the public were produced by “Process 2” E. coli plasmid DNA manufacture, which has led to plasmid DNA contamination of vaccine vials. 94
Beyond any clinical trial data and the process required to obtain initial approval from regulatory authorities, is the absolutely vital need to recognise that all therapeutic agents must be continuously monitored and subject to the red flags of vigilant surveillance.
Lack of recognition of pharmacovigilance data
Historic precedence in pharmacovigilance, safety and product recall has not been followed with respect to the COVID-19 gene-based vaccines, as shown by reports on https://www.vaersanalysis.info/ which collates weekly updates of data from the US CDC’s Vaccine Adverse Event Reporting System (VAERS) (Figure 4). The methodology used by vaersanalysis.info is presented in the supplemental materials. Reported suspected deaths for major drug/vaccine recalls versus COVID-19 vaccine reported suspected deaths. From VAERS Analysis
43
(with permission).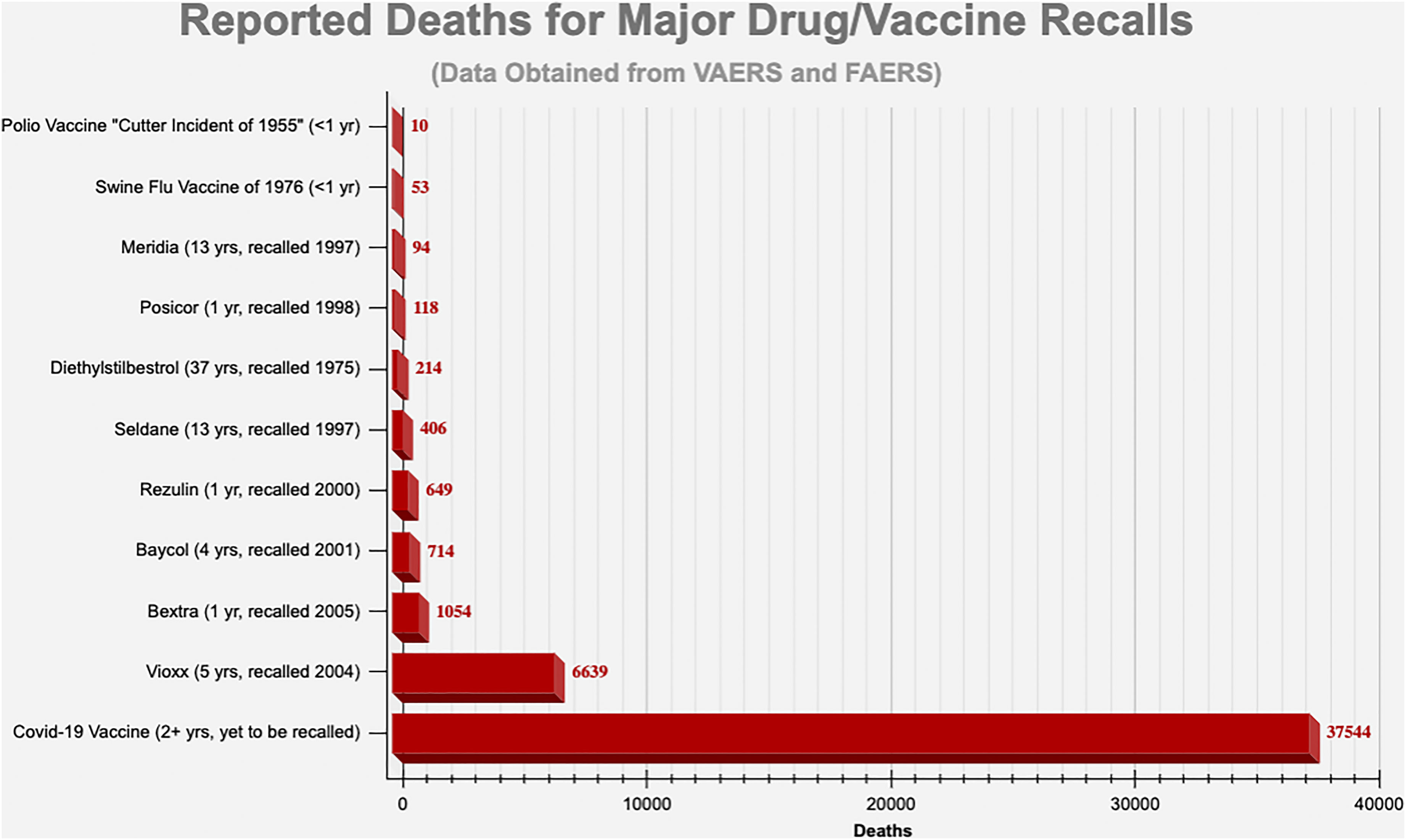
A polio vaccine was withdrawn after just 10 death reports, 95 the Swine Flu vaccine of 1976 was recalled after just 25 of the ultimate 53 death reports. 96
Not only are adverse events exceedingly high for the COVID-19 vaccines compared to all other vaccines (Figure 1), but deaths related to vaccines based per million doses show an unprecedented performance for the COVID-19 gene-based agents. Comparison with the influenza vaccine for which more doses have been dispensed is noteworthy (Figure 5). Suspected deaths per million doses of vaccine. Distributed doses in millions. Traditional versus COVID-19 vaccines. From: VAERS Analysis
43
(with permission). See also supplementary materials for further information on this graph.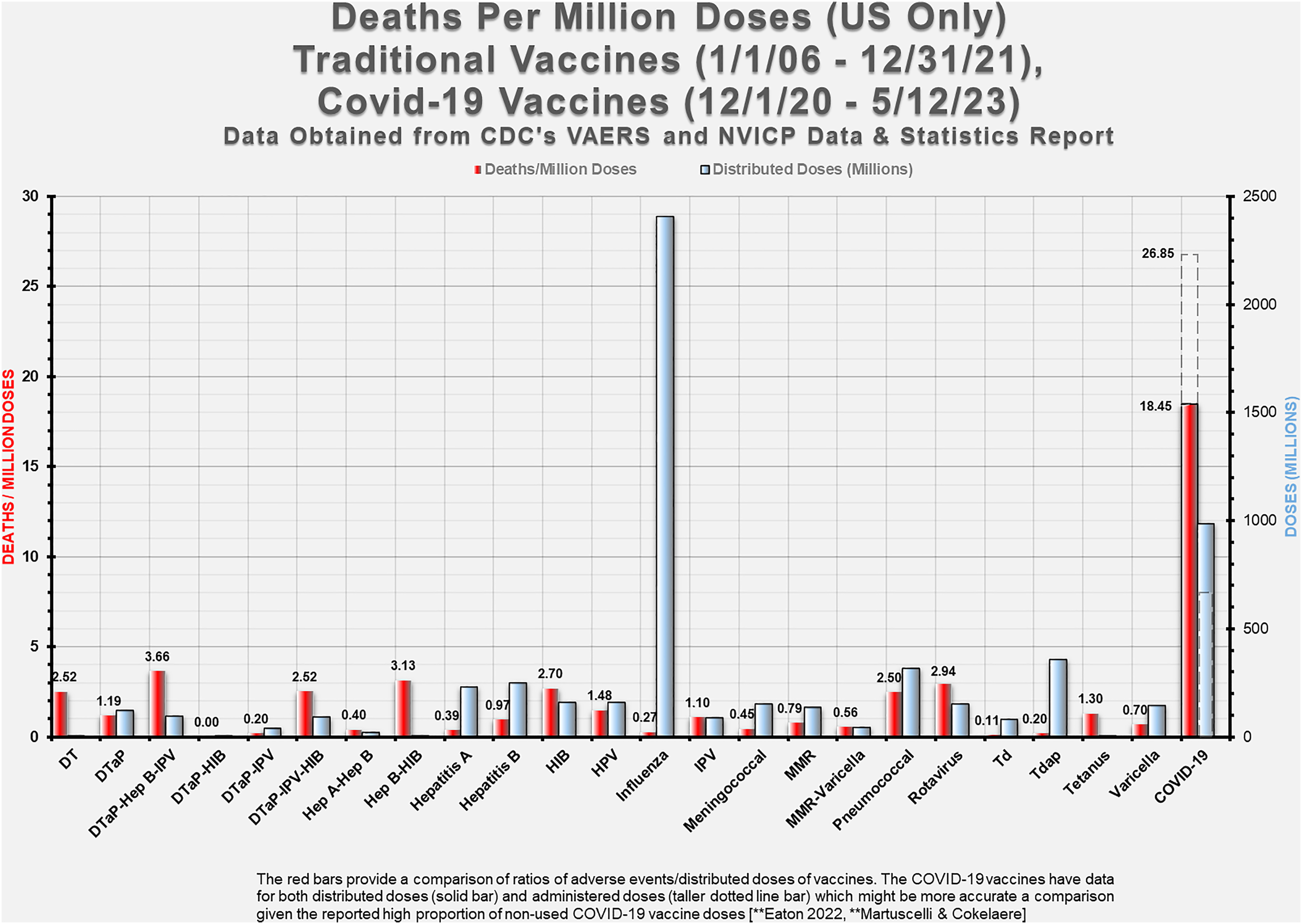
The red bars provide a comparison of ratios of adverse events/distributed doses of vaccines. The COVID-19 vaccines have data for both distributed doses (solid bar) and administered doses (taller dotted line bar) which might be a more accurate comparison given the reported high proportion of non-used COVID-19 vaccine doses.97,98
Pharmacovigilance underestimation factor
Vioxx data suggests the FDA’s adverse event database (FAERS) underestimates deaths by a factor of 5- to 9-fold.88,99 With deaths from strokes added to heart attacks, the under-estimation factor is likely to have been greater. 24 Yet, since the advent of the COVID-19 vaccines, health authorities have strenuously suggested the unprecedented adverse events are over-reported and thus overestimated. For example, the Australian Therapeutics Goods Administration (TGA) claim of overestimation by its passive system Database of Adverse Event Notifications (DAEN) is directly contradicted by the Australian National Centre for Immunisation Research and Surveillance (NCIRS), who operate the active prompted submissions to the AusVaxSafety database. While active AusVaxSafety data for Pfizer, 100 Moderna, 101 and AstraZeneca 102 vaccines failed to question around severe adverse events, and is thus incomplete, it still reflects far greater numbers of adverse events than the passively collected TGA DAEN figures.
In the US, government quality assurance suggests that the CDC’s VAERS under-reports by a factor of 10- to 100-fold – that only 1%–10% of all serious vaccine injuries are recorded. 103 VAERS sensitivity to capture serious adverse events well-known to be caused by vaccines, namely anaphylaxis and Guillain-Barré syndrome, ranged from 12% to 76%, but mostly around 25% for several vaccines. In other words, an underestimation factor of 4-fold. 104
These pharmacovigilance databases err decidedly on the side of underestimation, not overestimation.
In this context, the TGA confirms 14 of 1004 deaths (to 29 October, 2023) reported as potentially associated with the COVID-19 vaccines authorised in Australia, 105 which implies the other 990 deaths (98.6%) reported, mostly by clinicians, are attributable to an overestimation factor. The TGA dismissal of the severity of its own DAEN data is at odds with all prior research and with the active surveillance systems.
The active surveillance AusVaxSafety survey data showed a dose response effect of increased mRNA in the higher ratio of adverse events from Moderna than Pfizer COVID-19 vaccines and in the higher rate after the second dose that follows soon after the first. Graphical representations of the statistics reveal high rates of “missing work, study or routine duties”. A graph from the AusVaxSafety survey for the Moderna vaccine
101
is presented in Figure 6. AusVaxSafety had a limited range of adverse events typical of reactogenicity to vaccines for respondents to select. Inability to perform normal activities is generally considered a criterion for serious adverse events, even though the survey did not specifically list them. Impact on routine activities of Moderna doses 1, 2 and booster. AusVaxSafety data as January 26, 2023.
101
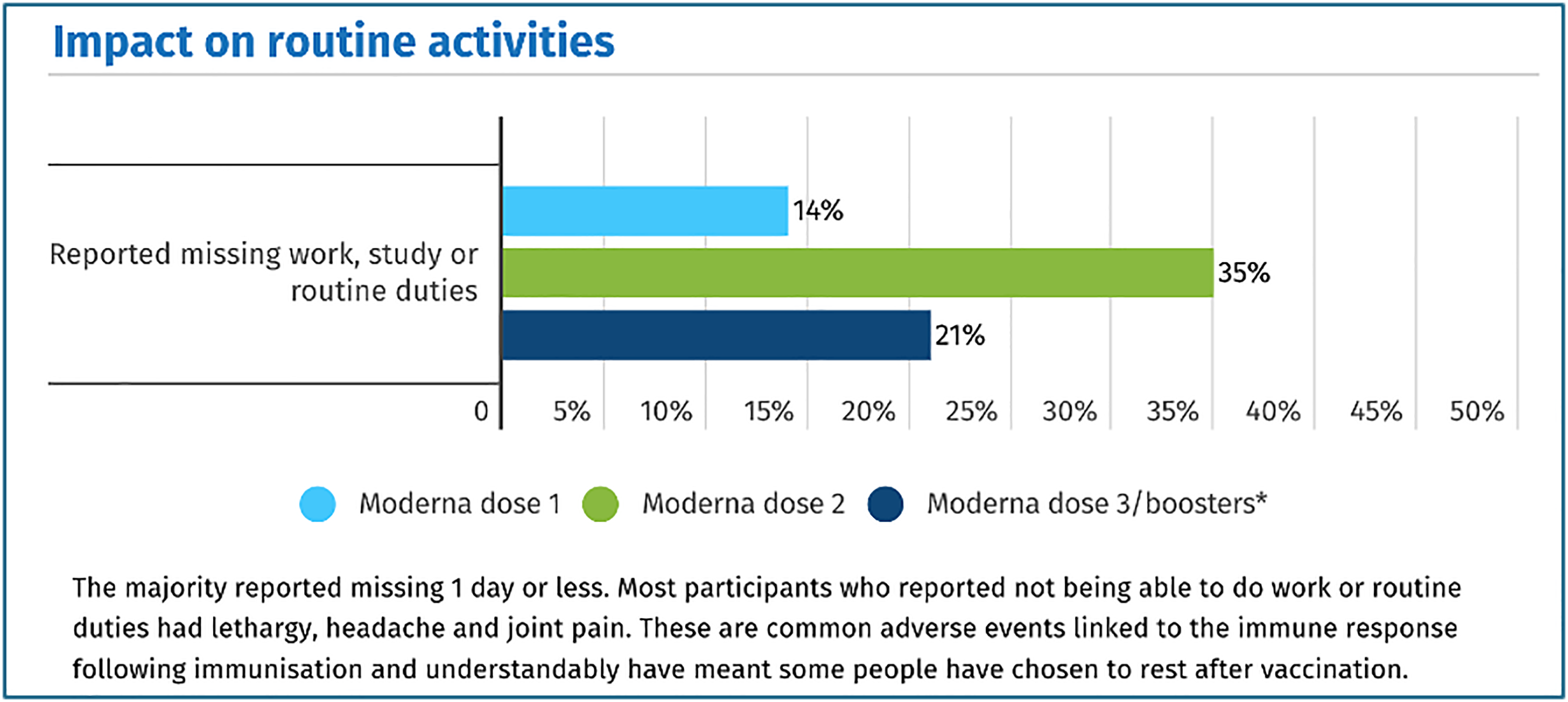
Educated hesitancy has been mocked. Figure 7 from the VAERS analysis data shows that the rate of adverse events per vaccine dose reported did not vary substantially across the age range. This contrasts with the severity of COVID-19 viral illness which was a relatively mild illness for younger age cohorts. Age stratification of adverse events after COVID-19 vaccination. From: VAERS Analysis
43
(with permission).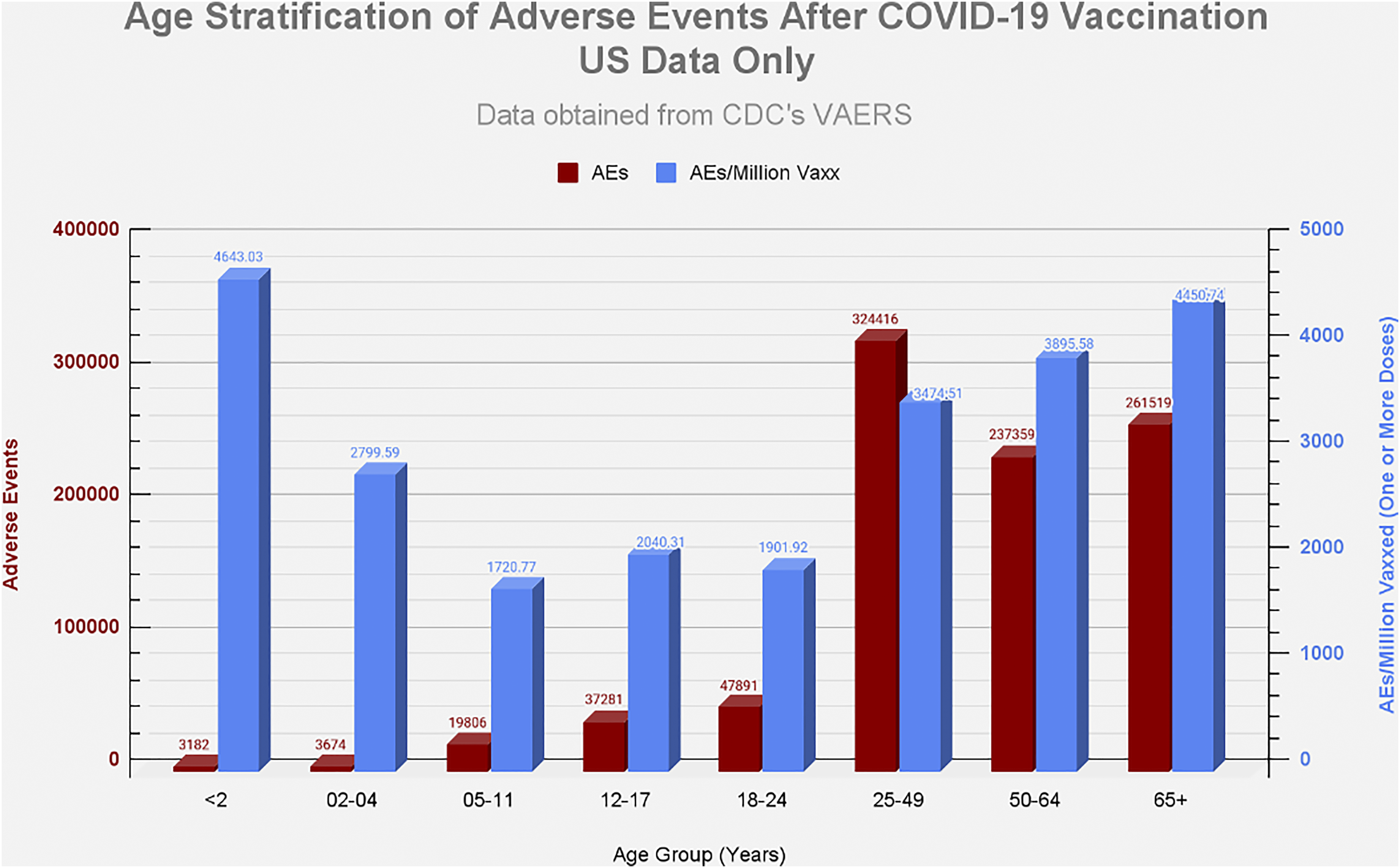
Pharmacovigilance and the future
Broadly, all pharmaceutical products are continuously experimental, observed and tracked by pharmacovigilance systems worldwide. The population ultimately becomes the long-term experiment. 1
Gene-based medicine in blanket form, with mass production at extremely low cost, is expected to become a significant market trend. 106 With the many gene-based therapeutic technologies planned, a vast new era of pathology may lie ahead.
Time honoured medical ethics and the precautionary principle must be reasserted. Commercial pressure, distortion of evidence base, authority bias and groupthink bureaucratic lockstep policy, all mitigate against cautious, safe-practice medical science.
Access to raw data, open discussion, freedom from censorship and heightened, active pharmacovigilance must be nurtured, if the health of humanity is to be better protected and if trust in the medical profession is to be fully restored.
Limitations
In this selective narrative review, limitations are embodied in the very nature of our subject matter – an exploration of conflicts between scientific integrity, data transparency and timely action on pharmacovigilance and adverse events, versus corporate ambitions to advertise, compete and market pharmaceutical agents for financial gain. The authors acknowledge limitations of free access to confidential data, a reliance upon Freedom of Information requests (themselves dependent upon the time, will and energies of interested parties), and of course dependency upon peer reviewed medical literature, an uncertain proportion of which has been shown to be unreliable, either because of exuberant optimism, publication haste or by deliberate design.25–28,30,32–35,57–74
The methodology for the graphs from https://www.vaersanalysis.info/ used in this paper, and limitations in the raw data used to compile those graphs, are described in the supplemental materials.
Conclusion
The fullest context is one in which the pharmaceutical industry has provided many remarkable drugs for the benefit of humanity. From this backdrop, we have selected a few of the most significant events in pharmaceutical recall history, in which commercial interest has dominated market strategy, and we have sought to derive key lessons from these.
A host of mechanisms are used by the pharmaceutical industry to promote and market their products. These include changes to the definitions or boundaries of disease, introduction of bias long before data collection begins, concealment of raw data, failure to collect safety data, or decisions not to report negative or unfavourable results.33,89,90
Gene codes for foreign protein production throughout the body are particularly novel. Close attention to pharmacovigilance data is imperative. Failure to withdraw the gene-based COVID-19 vaccines from the market, despite clear indications of harms, is not without precedent – as has been seen with Merck’s Vioxx (rofecoxib).
Excess mortality figures are high at present in many countries that have deployed the novel and experimental gene-based COVID-19 vaccines. As open-mindedness, objectivity and curiosity are essential to good science, we must immediately include new corporate products in our discussions about excess mortality and its possible causes. Drug recalls have been significant and numerous over recent decades. It may well be high time for the recall of still more.
Supplemental Material
Supplemental Material - Pharmaceutical product recall and educated hesitancy towards new drugs and novel vaccines
Supplemental Material for Pharmaceutical product recall and educated hesitancy towards new drugs and novel vaccines by Peter Rhodes and Peter I Parry in International Journal of Risk & Safety in Medicine.
Supplemental Material
Supplemental Material - Pharmaceutical product recall and educated hesitancy towards new drugs and novel vaccines
Supplemental Material for Pharmaceutical product recall and educated hesitancy towards new drugs and novel vaccines by Peter Rhodes and Peter I Parry in International Journal of Risk & Safety in Medicine.
Footnotes
Acknowledgements
The authors acknowledge the correspondence and assistance of the statistician at https://www.vaersanalysis.info/ in explaining the methodology (![]() ) for the VAERS pharmacovigilance data used in their charts and gratitude for permission to use in our paper.
) for the VAERS pharmacovigilance data used in their charts and gratitude for permission to use in our paper.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.