
Abstract
Immunoglobulin G (IgG) is a commonly used treatment for chronic neuromuscular disorders (NMDs), such as chronic inflammatory demyelinating polyneuropathy and multifocal motor neuropathy. IgG therapy has also shown promise in treating other NMDs including myasthenia gravis, polymyositis, and dermatomyositis. IgG is administered as either intravenous immunoglobulin (IVIg) or subcutaneous immunoglobulin (SCIg), with SCIg use becoming more popular due to the treatment burden associated with IVIg. IVIg requires regular venous access; long infusions (typically 4-6 hours); and can result in systemic adverse events (AEs) for some patients. In contrast, SCIg can be self-administered at home with shorter infusions (approximately 1 hour) and fewer systemic AEs. As patient care shifts toward home-based settings, the role of the pharmacist is paramount in providing a continuation of care and acting as the bridge between patient and clinic. Pharmacists with a good understanding of current recommendations, dosing strategies, and administration routes for IgG therapy are best placed to support patients. The aims of this review are to highlight the evidence supporting IgG therapy in the treatment of NMDs and provide practical information on patient management and IVIg/SCIg dosing in order to guide pharmacists on optimizing clinical outcomes and patient care.
Keywords
Introduction
Chronic autoimmune neuromuscular disorders (NMDs) can be broadly classified as neuropathies, myopathies, and neuromuscular junction disorders. 1 Clinical manifestations of NMDs are wide-ranging, but muscular weakness is experienced by most patients. This weakness may be progressive in nature and can have a significant impact on physical function and quality of life (QOL).1,2
Intravenous immunoglobulin (IVIg) is a mainstay of therapy for many patients with chronic autoimmune NMDs, including chronic inflammatory demyelinating polyneuropathy (CIDP); multifocal motor neuropathy (MMN); myasthenia gravis (MG); polymyositis (PM); and dermatomyositis (DM).3-6 Historically, immunoglobulin G (IgG) therapy has been administered via the intravenous route with a range of products available, with different purification, stabilization, and virus inactivation methods. 7 However, subcutaneous immunoglobulin (SCIg) has seen an increasing interest in recent years, as it removes the need for regular venous access and can allow patients to self-infuse at a location of their choice, which may be easier for patients with mobility issues, and without assistance from a health care professional (HCP).8,9 Subcutaneous doses are absorbed into the intravascular compartment slowly, which can result in fewer systemic adverse events (AEs) potentially eliminating the need to premedicate.8,9 The Food and Drug Administration (FDA) approved the first IgG formulation designed specifically for subcutaneous injection in 2006 for primary immune deficiency diseases (PIDD) since then further SCIg formulations and indications for use have also been approved. 10
SCIg therapy is widely used by patients with PIDD and pharmacists should be familiar with dosing and transitioning patients from IVIg. However, it is important to note that the doses, infusion parameters, and requirements for dose conversion during the transition from IVIg to SCIg differ for PIDD compared with neurological disorders, with higher doses generally used in NMDs (Table 1). 8 IgPro20 (Hizentra, a 20% subcutaneous immunoglobulin solution) was approved by the FDA for maintenance treatment of CIDP in 2018 based on the Polyneuropathy and Treatment with Hizentra (PATH) study, a dose-ranging phase III randomized controlled trial (RCT), which showed that SCIg reduced the risk of relapse or study withdrawal for any reason versus placebo in patients with CIDP. 11 While SCIg is less established in chronic NMDs, studies investigating SCIg use have reported positive results.12-16 A meta-analysis of studies comparing SCIg with IVIg in CIDP and MMN concluded that SCIg is as effective as IVIg in preventing muscle strength deterioration. 17 Additionally, the risk of moderate–severe AEs was reduced by 28%. 17 There is also rising evidence that SCIg is effective and well tolerated in patients with other neurological disorders, such as MG and myositis.18-22
Summary of Current IgG Treatment Guidelines for PIDD and Chronic Autoimmune NMDs.

Abbreviations: AAN, American Academy of Neurology; AANEM, American Association of Neuromuscular and Electrodiagnostic Medicine; bw, bodyweight; CIDP, chronic inflammatory demyelinating polyneuropathy; DM, dermatomyositis; EFNS, European Federation of Neurological Societies; FDA, food and drug administration; GBS, Guillain-Barré syndrome; IgG, immunoglobulin G; IVIg, intravenous immunoglobulin; MG, myasthenia gravis; MMN, multifocal motor neuropathy; NA, not applicable; PIDD, primary immunodeficiency diseases; PLEX, plasma exchange; PM, polymyositis; PNS, Peripheral Nerve Society; RCT, randomized controlled trials; SCIg; subcutaneous immunoglobulin.
a Guidelines issued by EFNS only.
Patients with chronic NMDs are more commonly opting to receive their care away from the clinic and choosing self-administration.17,23 Moreover, insurance providers, accountable care organizations, and pharmacy benefit managers are helping to drive this trend with the site of care initiatives and by encouraging SCIg where appropriate. 24 As a result, there is a growing need for pharmacists to have a good understanding of current recommendations and new approvals. Here, we collate the current guidelines and patient management recommendations, including monitoring and the use of evidence-based disability scales. The aims of this article are to (1) increase awareness of the role of IgG therapy in the treatment of NMDs, (2) provide practical information on dosing with IVIg and SCIg and patient management, and (3) encourage pharmacist involvement and act as a guide on strategies for navigating AEs with IgG therapy.
Disease Management in Chronic NMDS With IGG Therapy
The literature provides a strong evidence base to support the use of IVIg for the treatment of chronic NMDs, and treatment guidelines recommend routine use of this treatment option.5,6,25,26 More recent studies report investigations of SCIg in NMDs. SCIg is in the FDA label for CIDP, but not mentioned in the published NMD recommendations that are provided in Table 1, likely relating to the fact that all relevant guidelines were published more than 6 years ago.
Management of CIDP
Background
CIDP is characterized by inflammation of the peripheral nervous system and nerve injury.1,27 Disease subtypes have different patterns of nerve involvement (eg, specific to sensory or motor nerves). 28 The pathophysiological mechanisms have not been fully elucidated, but autoimmune mechanisms are believed to play an important role. 27 CIDP disease course is variable, as patients may show gradual deterioration over time, stepwise deterioration, or experience periods of relapse and remission.27,28 The main clinical symptoms of CIDP are typically paresthesia (followed shortly by weakness) and numbness, which worsen over time. 28 Motor deficits are the most common symptom with patients experiencing weakness both distally (eg, hand weakness or foot drop) and proximally (eg, difficulty climbing stairs or lifting objects) and can also experience problems with fine finger control. 29 Patients may also have difficulty walking, often requiring mobility aids, such as wheelchairs.27,28 Common features of CIDP also include decreased sensation, ataxia, and less commonly neuropathic pain. 28 Overall, the long-term prognosis for CIDP patients is good. A review of several small-scale studies using a disability grade reported 73% to 87% of patients receiving treatment were still ambulatory with mild symptoms after 5- to 10-year follow-up.28,30,31 Furthermore, 2 studies reported that a quarter of patients achieved remission and no longer required treatment.32,33
Evidence for IgG therapy
The main treatment options for patients with CIDP are IgG therapy, corticosteroids, and plasma exchange. 27 Immunosuppressants, such as azathioprine or methotrexate, can be considered as an add-on therapy for patients not adequately responding to principal options. 34 IVIg is a well-established treatment for CIDP and there is evidence to support the use of SCIg in CIDP, with Hizentra now FDA-approved as maintenance therapy in this setting. 35 The PATH study, performed in 172 patients with CIDP with confirmed dependency on IVIg therapy (dose ≤1.6 g/kg every 4 weeks), was placebo-controlled and investigated 2 weekly doses of SCIg (0.2 g/kg and 0.4 g/kg, administered as 1 or 2 mL/kg, respectively). 11 The PATH primary outcome was the proportion of patients with a CIDP relapse or withdrawal from the study. 11 In the study, 63% (95% CI: 50-74) of patients on placebo, 39% (95% CI: 27-52) on 0.2 g/kg SCIg, and 33% (95% CI: 22-46) on 0.4 g/kg SCIg had CIDP relapse/withdrawal from the study. 11 When looking at CIDP relapse only, these figures were 56% (95% CI: 43.3-68.2) with placebo, 33% (95% CI: 22-46) with 0.2 g/kg SCIg, and 19% (95% CI: 11-31) with 0.4 g/kg SCIg. 11 Absolute risk reductions compared with placebo for CIDP relapse or study withdrawal for any other reason were 30% (95% CI: 12-46) with SCIg 0.4 g/kg and 25% (6%-41%) with SCIg 0.2 g/kg (both P < .01). 11 Both doses of SCIg were similarly well tolerated and the study concluded that, in clinical practice, the weekly dose of SCIg could be varied across the range 0.2 to 0.4 g/kg, depending on clinical requirements. 11 Furthermore, 88% of all patients enrolled in the PATH study reported that learning subcutaneous self-administration was easy. 11
In a smaller, single-arm study, 16 drug-naive patients with CIDP were initially treated with IVIg (0.4 g/kg/d for 5 consecutive days) before commencing SCIg treatment (0.4 g/kg/wk) after 4 weeks. 36 Long-term follow-up (at 12 and 24 months) demonstrated that SCIg as a first-line treatment improved nerve conduction and measures of muscle strength and disability in patients with CIDP. 36 A limitation of this study was that an excellent response to the initial IVIg dose could have persisted and affected the SCIg results at 12 or 24 months. 36 Moreover, a retrospective study in various NMDs, including CIDP, observed maintained clinical stabilization in the majority of patients with a mean follow-up of over 3 years. 37 These data support early and long-term treatment with SCIg; however, it should be noted that the initial time to clinical response can be longer with SCIg compared with IVIg. 38 Differences in time to response were observed in a small randomized, single-blind, cross-over study in 20 drug-naive patients with CIDP, where an IVIg infusion (0.4 g/kg/d over 5 days) was compared with SCIg (0.4 g/kg/wk for 5 weeks). 38 Both SCIg and IVIg improved motor performance to a similar degree, but with earlier maximal improvement following IVIg infusion; isokinetic muscle strength peaked 2 weeks after the IVIg infusion compared with a peak after 5 weeks of weekly SCIg infusions. 38
Management of MMN
Background
MMN is one of a small number of neuropathies that cause motor deficits without any sensory loss.39,40 The full pathogenesis of MMN is unknown; however, recent reviews have implicated immunological targeting of the nodes of Ranvier and paranodal regions, leading to persistent conduction block.41-43 The most commonly affected nerves are the ulnar, median, radial, and tibial nerves. 39 The principal symptoms of MMN relate to muscle weakness of the distal limbs and are likely to include impaired wrist or hand function.39,40 Symptoms are typically asymmetric and slowly progressive. 40 Criteria published by the European Federation of Neurological Societies/Peripheral Nerve Society in 2010 are often used to diagnose MMN. 44 However, with an initial clinical presentation similar to that of amyotrophic lateral sclerosis (ALS), diagnosis is potentially challenging. 40 Conversely, unlike ALS, life expectancy is normal in MMN. 39
Evidence for IgG therapy
Patients with MMN typically respond well to IgG therapy, while other treatments (eg, corticosteroids, plasma exchange) are ineffective, have insufficient evidence, or do not have a favorable risk–benefit profile.39,40,45 IVIg is the only approved treatment for MMN, but small studies have suggested that SCIg may be a viable alternative.12,23,46,47 In a short-term, randomized crossover study performed in 9 patients, SCIg given twice or thrice weekly (15.2 g [95 mL] to 24.8 g [155 mL] per week) showed favorable tolerability and similar efficacy as IVIg. 48 More recently, Katzberg et al evaluated SCIg in 15 patients with MMN, with weekly SCIg doses between 0.12 and 0.5 g/kg bodyweight dependent on previous IVIg dose. 49 Patients with MMN tolerated SCIg and maintained muscle strength, although some patients required closer monitoring as increasing weakness started to develop during the 6-month study. 49
Management of MG
Background
MG refers to a group of diseases where defective neuromuscular transmission leads to muscle weakness that is exacerbated by physical exertion.50,51 Weakness of the extrinsic ocular muscle is likely to be the first symptom of MG, and the disease often progresses to involve other muscles such as those of the neck and jaw. 52 MG has the potential to be life-threatening as it can cause dysphagia or respiratory failure (ie, myasthenic crisis).50,51 Antibodies targeting the nicotinic acetylcholine receptor (AchR), muscle-specific kinase (MuSK), or low-density lipoprotein receptor-related protein 4 (LRP4) are often present.1,51 These appear to play important pathophysiological roles, but a subset of MG patients are “triple seronegative.”50,51 Single-fiber electromyography and repetitive nerve stimulation tests are the predominant methods of diagnosing MG, while immunological tests for the antibodies mentioned above are supportive of a positive diagnosis. 50
Evidence for IgG therapy
Most patients with MG are treated with acetylcholinesterase inhibitors, together with corticosteroids or immunosuppressants if required, and specific therapies for patients with refractory disease, myasthenic crisis, or thymomas.50,51,53 A Cochrane review of IVIg in MG, published in 2012, concluded that although the RCTs in MG were small and underpowered, there was evidence suggesting IVIg was similarly effective as other treatment options for progressive MG or exacerbations. 54 A subsequent review highlighted positive evidence for IVIg treatment of patients with worsening MG or myasthenic crisis, although currently only retrospective studies support IVIg for long-term maintenance therapy. 55 However, a small prospective study found IVIg administered as a loading dose of 2 g/kg, followed by booster doses of 0.4 g/kg every 4 to 12 weeks, resulted in a persistent decline of 50% in the quantitative myasthenia gravis score (QMGS, a marker of disease severity) after 24-month follow-up. 56 A retrospective Canadian study initiated or switched 9 patients with MG to weekly SCIg (0.29-0.39 g/kg bodyweight); of these, 3 were IgG naive and 6 were receiving IVIg. 57 The study found statistically significant improvements in QOL, with no reported exacerbations after starting SCIg therapy and no severe SCIg-related complications. 57 SCIg has also been investigated in MG in an open-label, phase III study, performed in patients with mild–moderate MG exacerbation. 18 Patients dosed with 2 g/kg 20% SCIg solution infused subcutaneously over a 4-week treatment period in a dose-escalating manner, reported that SCIg decreased QMGS from 14.9 ± 4.1 to 9.8 ± 5.6 (P < .0001) and was well tolerated. 18 Moreover, an ongoing study of SCIg in MG has reported promising results from an interim analysis. 58
Management of PM and DM
Background
PM and DM are 2 types of chronic inflammatory disorders characterized by muscle inflammation and weakness mainly affecting proximal skeletal muscles. 59 DM is distinguishable from PM primarily by the occurrence of skin abnormalities and rashes. 59 DM skin rashes can occur over the face (particularly the eyelids), neck, chest, shoulders, and upper back or around the joints, typically elbows, knees, and ankles. 59 This rash tends to be violet-colored, can be itchy and painful, and is often aggravated by sun exposure. 60 PM and DM are autoimmune in nature and are associated with increased morbidity and mortality often as a result of severe muscle weakness or visceral organ involvement. 59 Both inflammatory myopathies can occur in pediatric populations, most commonly as juvenile dermatomyositis (JDM). 61 Most cases of JDM start at ages 5 to 10 years, with girls affected around twice as often as boys. 62
Evidence for IgG Therapy
Myositis is usually treated with corticosteroids or immunosuppressant drugs. IgG therapy is recommended as add-on therapy by the Myositis Association in refractory myositis or in patients with comorbidities.5,60 Case reports have described benefits following IVIg treatment in patients with esophageal involvement and pulmonary complications.63,64 A 63-year-old man with interstitial lung disease associated with PM/DM was reported to have 5 months of progressive dyspnea and weakness. 63 After 3 monthly doses of IVIg at 2 g/kg, he sustained clinical remission for more than 2 years, with clinical markers of disease such as creatine kinase levels returning to normal levels and lung function measures significantly improving. 63 In a retrospective analysis of 73 patients with PM or DM, with steroid-resistant esophageal involvement, treatment with IVIg (2 g/kg) resolved esophageal symptoms (dysphagia, coughing while eating, and gastroesophageal reflux into the pharynx and/or mouth) in 82.2% of patients, eliminating the need for enteral feeding tubes. 64 In JDM, a retrospective study of 18 pediatric patients treated with steroids demonstrated that IVIg treatment led to clinical improvement in 12 patients and was associated with a >50% reduction in corticosteroid therapy. 65 A case study reporting on the use of SCIg in a patient with PM concluded that switching from IVIg (2 g/kg per month) to SCIg (1.3 g/kg per month or approx. 0.33 g/kg per week) was associated with increased QOL and significant improvements in treatment satisfaction with only mild local reactions observed. 19 A study has reported on the use of SCIg in 7 patients with active and refractory PM or DM. SCIg was administered weekly (0.5 g/kg bodyweight) and all patients showed favorable clinical responses, no relapse of disease, and improvement of short-form 36 health survey (SF-36) QOL scores (with the highest scores being seen in global mental health), demonstrating the beneficial effects of SCIg, with good tolerability and safety. 21
Safety With IGG Therapy
IVIg
IVIg is considered to be one of the first-line treatments for CIDP and MMN. Although this treatment is generally well-tolerated, there are some important safety considerations. Some AEs can occur immediately upon treatment, while others have delayed onset. Most AEs with IVIg are mild and transient; these may include headache, nausea, and flu-like symptoms (eg, fatigue, fever). 66 Headache has been reported to be the most common systemic AE associated with the administration of IVIg, occurring at a frequency of 5% to 20% of IVIg infusions.11,67 Mild–moderate AEs can often be controlled by medications administered before or after infusion or by reducing the infusion rate to the maximum tolerated. Additionally, HCPs should ensure adequate hydration before and during the IV administration (Table 2). 67
Management of AEs Associated With IVIg Therapy.

Abbreviations: AEs, adverse events; IgG, immunoglobulin G; IVIg, intravenous immunoglobulin; NSAIDs, nonsteroidal anti-inflammatory drugs; SCIg, subcutaneous immunoglobulin.
Although uncommon, some serious AEs have been reported with IVIg. These include aseptic meningitis, thromboembolic events (TEEs), cardiac events, renal impairment, hemolysis, gastrointestinal problems, and transfusion-related acute lung injury (TRALI).66,67 Over prolonged periods of time, patients receive large numbers of infusions, meaning that a significant proportion of patients may at some time experience one of these AEs.
Aseptic meningitis
Up to 1% of IVIg patients can be affected by aseptic meningitis, with symptoms (eg, persistent headache, nausea, vomiting, photophobia, fever) likely to appear within 48 hours of IVIg infusion.66,68,69 Although headache is a common side effect of IVIg therapy, aseptic meningitis is more likely to occur in individuals with a history of migraines, and it should be suspected in all patients with long-lasting headaches following IVIg despite appropriate premedication. 66 Monitoring should include how long headaches persist post-IV infusion.
Thromboembolic events
In a retrospective analysis performed in patients with CIDP or MMN, a daily IVIg dose of 35 g or higher was shown to increase the risk of TEEs. 70 The authors of this study considered a range of patient characteristics to be risk factors for TEEs, including male gender, age over 60 years, lack of ability to walk unaided, atrial fibrillation, dyslipidemia or family history of thromboembolic disease, or a diagnosed comorbidity such as diabetes, hypertension, or coronary disease. 70 A significantly higher mean number of these risk factors was present in patients who developed TEEs. 70 Pretreatment screening for TEE history and comorbidities can provide early signals that can be useful when monitoring potential problems over time.
Renal dysfunction
The risk of renal dysfunction has been linked with the use of stabilizers (particularly sucrose) in IVIg formulations. 71 Renal failure is most likely to occur in patients with preexisting conditions such as renal insufficiency or diabetes. In these patients, sucrose-stabilized IVIg formulations (Carimune—discontinued in 2018 and Tegeline—available in Europe and Latin America contain sucrose) should be avoided as these products are associated with a higher risk of renal failure than non-sucrose stabilized formulations. 71
Hemolysis
The risk of hemolysis is increased in patients receiving high doses of IVIg, those receiving IVIg for inflammatory or autoimmune disorders, and in patients with non-O blood groups.72,73 Monitoring hemoglobin levels closely in all patients receiving high-dose IVIg therapy is recommended. However, the risk of this complication can depend on the quantity of isohemagglutinins present in the IgG product relative to the patient’s own blood group. 74 It may be possible to reduce the risk by lowering levels of isohemagglutinins during the manufacture of IgG products: To this end, immunoaffinity chromatography is performed during production of Privigen and Hizentra. 74
SCIg
The subcutaneous route of administration delivers IgG gradually into the intravascular compartment, resulting in peak plasma concentrations that are approximately 40% lower than after intravenous administration. 75 Consequently, the lower peak plasma values with SCIg may contribute to the tolerability differences between SCIg and IVIg. 26 Several studies have reported lower AE incidence or severity in patients with NMDs receiving SCIg versus IVIg. 17 In an open-label Danish study involving 86 patients, the severity of headache and nausea was significantly lower with SCIg (P < .0001 for both). 76 In a subsequent study of 23 patients, the same research group assessed hemolytic activity and reported that a switch from IVIg to SCIg led to statistically significant improvements in relevant laboratory variables. 77 Furthermore, a meta-analysis including 138 patients across 8 studies reported that the relative risk of moderate to severe AEs was 28% lower with SCIg versus IVIg (95% CI: 0.11-0.76). 17 Local injection site reactions (eg, swelling, redness, itching) are the most common AEs with SCIg.78,79 Such AEs are usually considered to be mild; their severity tends to reduce over time as the number of infusions increases, and they can be mitigated by ensuring that infusion technique is optimal. 79 Considering that SCIg is usually self-administered, it is important that patients are prepared for the possible occurrence of AEs as part of their treatment training and that telephone support is available if needed (particularly during the first few home treatments). 79
Treatment Burden With IGG Therapy
Long-term IVIg treatment requires regular venous access, which can be uncomfortable and invasive for the patient. Identification of suitable veins for infusing the large volumes that are required in chronic NMDs becomes more challenging the longer the patient remains on regular treatment. Implantation of a central venous line or port is an option for individuals with poor venous access. This approach is effective in simplifying the infusion procedure, but it also increases the risks of certain complications such as vascular injury, infections, and a further increased risk of TEEs.80,81 One study reported complications in up to 40% of patients following catheter placement. 82 The associated complications with venous access devices can result in increased health care costs. For example, catheter-related bloodstream infections have a reported incidence between 0.177 and 0.270 infections per 1000 catheter days, estimated to be an additional average cost per infection of US$5000 to US$34 000. 83
The therapeutic effect of IVIg may begin to reduce toward the end of the typical 3- to 4-week interval between IVIg treatments, which can result in “wear-off” symptoms between doses, such as a return of muscle weakness and fatigue. 84 Compared with IVIg, SCIg involves the administration of smaller doses more frequently, resulting in more stable serum IgG levels with smaller variations between peaks and troughs which may reduce the likelihood of treatment-related fluctuations, including adverse events and improve the maintenance of functional ability.78,85,86
Opportunities for the Pharmacist
As more patients with chronic autoimmune NMDs become involved in their own disease management, the need for support in the home setting is growing. Provision of guidance for optimal administration of SCIg or IVIg is an important part of this support, particularly during the transition to home-based therapy.
Dose Adjustments With SCIg
The IgG dose and volume of administration required to treat PIDD compared with NMDs differs. IgG therapies are typically available as 10%, 16%, or 20% preparations. Guidelines for IVIg treatment of PIDD recommend a starting dose of 0.4 to 0.6 g/kg every 3 to 4 weeks; SCIg is used at a starting dose of 0.1 to 0.2 g/kg/wk (Table 1). 87 In contrast, guidelines suggest IVIg dosing for most NMDs commence with a 2 g/kg loading dose over 2 to 5 days, followed by a maintenance dose of 1 g/kg every 2 to 4 weeks. 87 Currently, there is no guidance recommending SCIg doses for NMDs; however, recent clinical studies have used loading doses of 0.2 g/kg/d for 5 days, followed by weekly maintenance doses of 0.2 to 0.4 g/kg using infusion volumes of 1 to 2 mL/kg, respectively.11,36 Similar to IVIg therapy, SCIg administration should be individualized for each patient. When transitioning patients from IVIg to SCIg therapy in PIDD, the FDA-recommends using a dose adjustment coefficient (DAC) of 1.37 to 1.53 depending on the SCIg percentage formulation (eg, SCIg dose = IVIg dose (g) × DAC/number of weeks between doses).35,87 There is currently no recommendation for a DAC when transitioning patients with NMDs and most studies to date have used the weekly equivalent of a 1:1 conversion from the previous IVIg dose.12,14,16,21 However, further dose adjustments may be required following the transition to optimize therapeutic benefit based on individual patient need (see Table 3 for a list of recent SCIg studies with dosing regimen).
SCIg Studies in CIDP, MMN, MG, and DM/PM With Sample Size, Dosing, Time Frame, and Outcome.
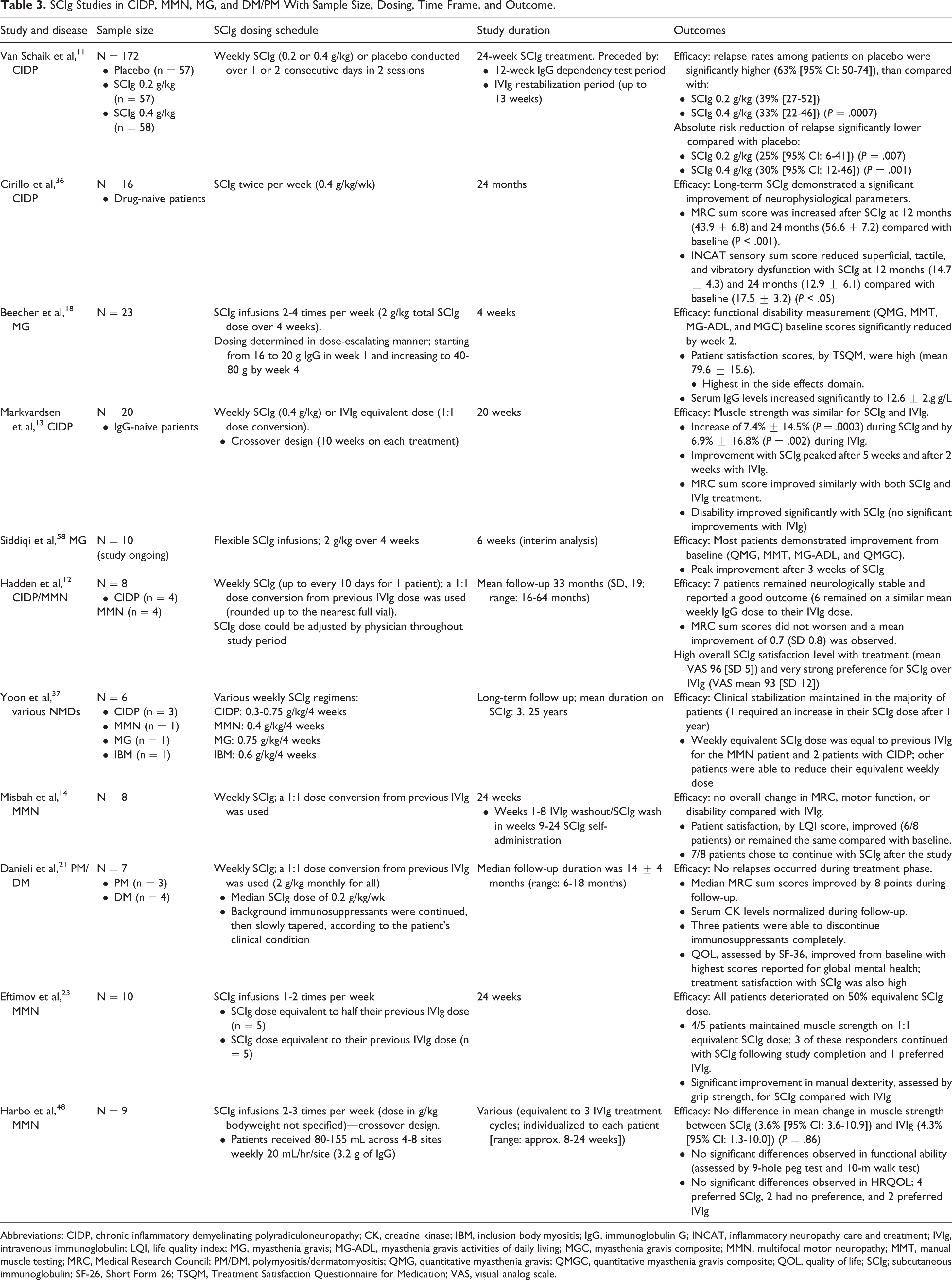
Abbreviations: CIDP, chronic inflammatory demyelinating polyradiculoneuropathy; CK, creatine kinase; IBM, inclusion body myositis; IgG, immunoglobulin G; INCAT, inflammatory neuropathy care and treatment; IVIg, intravenous immunoglobulin; LQI, life quality index; MG, myasthenia gravis; MG-ADL, myasthenia gravis activities of daily living; MGC, myasthenia gravis composite; MMN, multifocal motor neuropathy; MMT, manual muscle testing; MRC, Medical Research Council; PM/DM, polymyositis/dermatomyositis; QMG, quantitative myasthenia gravis; QMGC, quantitative myasthenia gravis composite; QOL, quality of life; SCIg; subcutaneous immunoglobulin; SF-26, Short Form 26; TSQM, Treatment Satisfaction Questionnaire for Medication; VAS, visual analog scale.
Optimizing Patient Management
Pharmacists have the opportunity to provide counseling and medication reconciliation as well as navigate adverse effects and patient monitoring; however, pharmacists are currently estimated to be underused in this regard. 88 Pharmacists may help monitor IgG treatment outcomes while using their patient-counseling skills to encourage treatment adherence, through direct consultation- or education-related activities. As shown in Table 4, a number of tools are available to help monitor NMD patients’ progress including mobility/functional tests (eg, grip strength; Medical Research Council muscle strength scale) and patient-reported questionnaires (eg, inflammatory neuropathy cause and treatment disability scale; inflammatory Rasch-built overall disability scale; and SF-36).89-91 Disease-specific tools can also be used, for example, MG-DIS and MG-QOL are MG-specific tools to assess disability and QOL. 92 All patients with chronic autoimmune NMDs should be monitored regularly and assessment data tracked over time to monitor patient/disease progression or improvement. Monitoring tools also provide payers and providers clinical information to justify keeping patients on service and treatment. Information collected should be shared with the physician, to provide patient status updates in-between clinic visits. If the patient is self-administering at home, the pharmacist can be best placed to collate this assessment data and collaborate with other care providers and the physician. Establishing a good relationship between providers and pharmacists, irrespective of infusion setting, should be the standard of care. Ultimately, however, the patient’s physician is usually responsible for confirming IgG dependence during follow-up visits, often to satisfy insurance providers.
Principal Tools for Assessing Status and Treatment Response in Patients With Chronic Autoimmune Neuromuscular Disorders.

Abbreviations: CMAS, Childhood Myositis Assessment Scale; CIDP, chronic inflammatory demyelinating polyneuropathy; DM, dermatomyositis; HAQ, health assessment questionnaire, INCAT, Inflammatory neuropathy cause and treatment; I-RODS, inflammatory Rasch-built overall disability scale; JDM, juvenile dermatomyositis; MG, myasthenia gravis; MGC, myasthenia gravis composite; MMN, multifocal motor neuropathy; MMT, manual muscle testing; MRC, medical research council; PM, polymyositis; SF-36, Short Form 36.
On-Going Patient Support
The dependency of patients with chronic autoimmune NMDs on IgG can fluctuate, with factors such as changes in weight, comorbidities, or lifestyle impacting the effectiveness and/or need for IgG treatment. Adjustments may be required to the IgG dose or infusion regimen. Pharmacists with the credentials and experience to provide direct patient care are well placed to assist patients with site selection, rotation, and potential issues/adverse reactions that could diminish tolerability and adherence. 93 Site rotation and condition is an essential check as patient adherence can lessen as discomfort increases. Table 5 provides some approaches for managing potential AEs with SCIg therapy. Additionally, in Table 6, there are several interventions that pharmacists can make to provide positive contributions to medication management, patient education, and counselling. 94
Management of AEs Associated With SCIg Therapy.

Abbreviations: AEs, adverse events; IVIg, intravenous immunoglobulin; NSAID, nonsteroidal anti-inflammatory drugs; SC, subcutaneous; SCIg, subcutaneous immunoglobulin.
Pharmacist Interventions and Their Possible Benefits. Nursing Staff May Contribute to (or lead) Some of the Interventions That Are Listed.

Abbreviations: IgG, immunoglobulin G; HCP, health care professional.
Cost Aspects of IGG Therapy
The cost of treating NMDs can be high, and the chronic nature of these diseases means therapy is often required long term. In a US review of health care usage in patients with CIDP, pharmacy cost was found to be the major cost driver accounting for 57% of total costs, of which IVIg accounted for 90%. 95 Several North American studies in patients with PIDD concluded that home-based SCIg resulted in reduced costs for the health care system compared with hospital-based IVIg.96-98 Moreover, increased productivity and reduced hospital-related absenteeism have been reported in patients with PIDD following a transition to SCIg.99,100 It anticipated that similar cost benefits will be seen in chronic NMDs.101,102
Conclusions
The use of IVIg therapy is the mainstay in effectively treating NMDs, but some patients have intolerable side effects and complications, and treatment can require inconvenient infusion durations and/or travel time when performed in a health care facility. SCIg is an alternative method of administration that can significantly reduce side effects and the need for premedication, giving patients more flexibility and control over their treatment planning. Currently, many pharmacists may be familiar with treatment protocols for PIDD but not NMDs. 87 By understanding the difference in doses, infusion parameters, and transition protocols involved in NMDs, pharmacists can play an important role in optimizing patients’ treatment. Furthermore, having access to the self-reporting and monitoring tools used by patients will help pharmacists support patients during their transition phase from hospital-based therapy to home-based SCIg, a transition which may ultimately lead to lower care costs.
In conclusion, pharmacists with knowledge of dosing strategies, potential side effects, and administration routes for IgG therapy can support NMD patients and act as a bridge between patient and clinic ensuring optimum patient care.
Footnotes
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article. E.M.T. reports non-financial support from CSL Behring during the preparation of this review manuscript; personal fees from Shire Pharmaceuticals, personal fees from Grifols Pharmaceuticals, non-financial support from CSL Behring outside of the submitted work. B.P. and D.D. have nothing to disclose.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Editorial support was provided by Meridian HealthComms Ltd funded by CSL Behring.