
Abstract
Peritoneal dialysis (PD) has important disadvantages compared to hemodialysis, including low plasma clearance and limited technique survival. A new device for sorbent-assisted (continuous flow) peritoneal dialysis (SAPD) has been designed that is based on continuous recirculation of peritoneal dialysate via a single-lumen peritoneal catheter with regeneration of spent dialysate by sorbents. SAPD treatment may enhance plasma clearance of uremic solutes by increasing the mass transfer area coefficient and maintenance of a high plasma-to-dialysate concentration gradient. In addition, SAPD treatment may preserve integrity of the peritoneal membrane for a longer period of time by avoiding the need for high initial glucose concentrations and by reducing the number of exchanges and (dis)connections of the peritoneal catheter, which may lower the risk of peritonitis. The primary aim of this first-in-human clinical trial is to evaluate the (short-term) clinical safety and performance of SAPD treatment in a small group (n = 12) of stable adult PD patients in a clinical setting (proof of concept). Key secondary objectives include an evaluation of efficacy in terms of plasma clearance, ultrafiltration, and patient tolerance.
This is a visual representation of the abstract.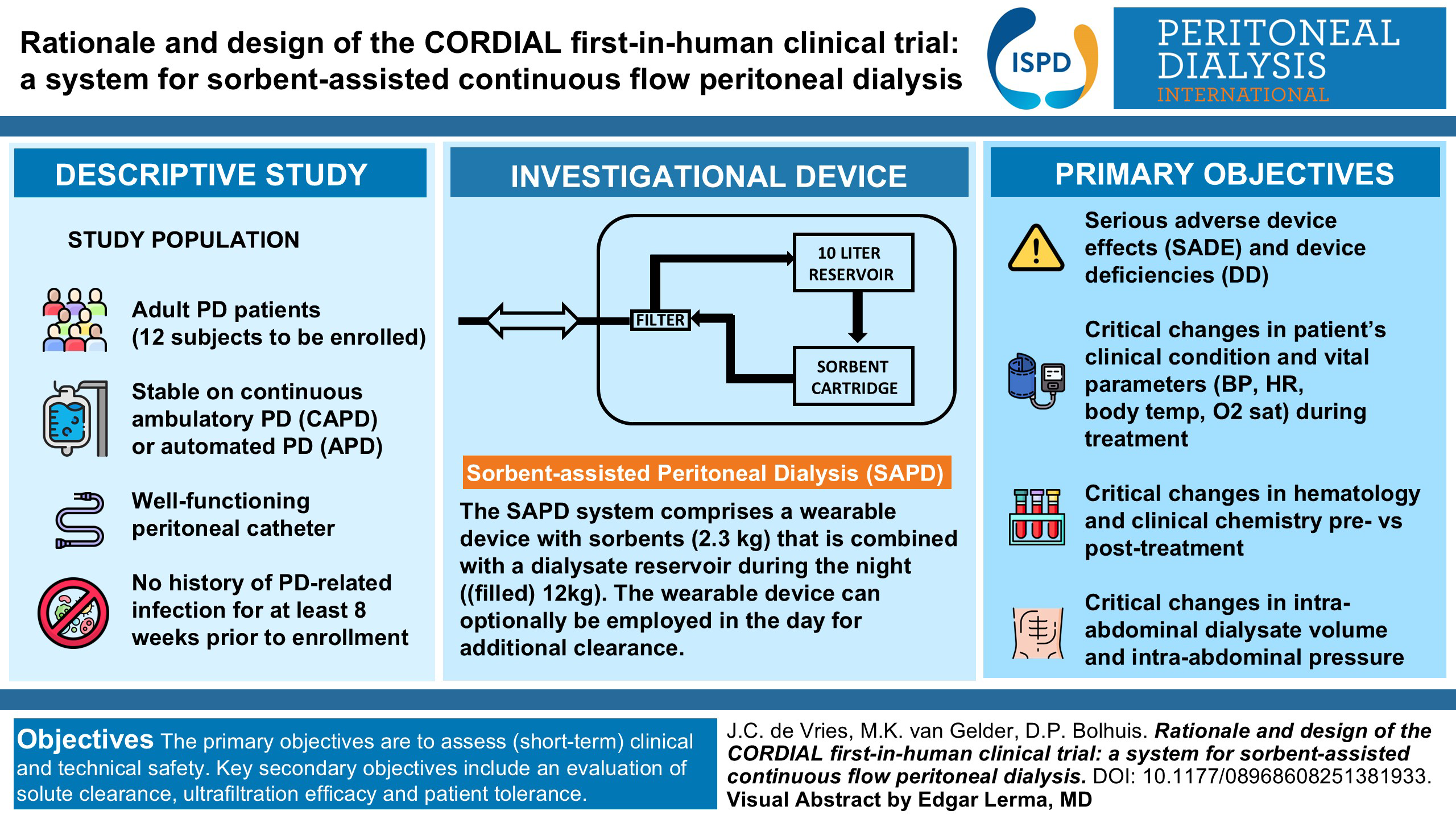
Rationale and background
Worldwide, the number of dialysis patients is 3.9 million of which 89% is treated with hemodialysis (HD) and 11% with peritoneal dialysis (PD). 1 PD offers several advantages compared with HD. It provides continuous and more gradual dialysis, resulting in more stable plasma solute concentrations and fluid status than with HD. It also provides more autonomy. Residual kidney function, associated with a survival benefit, is better preserved than with HD. 2 Costs for PD are generally lower than those for standard in-center HD. 3 However, conventional PD has important disadvantages. Uremic solute clearance is relatively low (lower than with HD) 4 and time on PD therapy is limited (median 3.7 years 5 ) primarily due to recurrent peritonitis6,7 and membrane failure. 8 Both recurrent peritonitis and chronic exposure of the peritoneal membrane to hypertonic glucose solutions cause pathological changes of the peritoneal membrane such as fibrosis and neoangiogenesis, and may eventually lead to ultrafiltration (UF) failure.6,8,9
To improve the existing shortcomings of conventional PD, a system for sorbent-assisted (continuous flow) peritoneal dialysis (SAPD) has been designed that introduces the following innovations: instead of refreshing peritoneal dialysate 4–6 times per day as performed with conventional PD, only one peritoneal dialysate filling is used that is continuously recirculated and regenerated by the SAPD system containing sorbents. This may improve plasma clearance in two ways: first, the continuous flow of dialysate may increase the mass transfer area coefficient (MTAC) of solutes across the peritoneal membrane as observed in patient studies with continuous flow PD,10–15 probably through reduction of diffusion resistances, renewal of stagnant fluid layers at the tissue surface and an increase of the effective membrane surface area. Second, regeneration of the dialysate will prevent its saturation with toxins, maintaining a high diffusion gradient across the peritoneal membrane that drives diffusive solute removal. Preliminary data in (pre)clinical trials, although scarce, suggest that SAPD is superior to conventional PD. However, the number of patients treated is limited, and many studies employ a setup using two peritoneal catheters (separate in- and outflow), while various SAPD devices have been designed for use in a single-lumen catheter setup. 15
Another benefit of SAPD treatment may be prolongation of time on PD therapy. By reducing the number of exchanges to one per day and therewith the number of (dis)connections to the PD catheter, SAPD treatment may lower peritonitis rates. 16 Furthermore, during SAPD treatment glucose is continuously released at a constant rate, maintaining the intraperitoneal glucose concentration at a minimum level for removing excess fluid, avoiding the need for high initial glucose concentrations as applied for static dwells, which may result in longer preservation of the peritoneal membrane integrity and reduce UF failure. In addition, UF efficiency may be improved. 17
The aim of this first-in-human clinical trial is to evaluate the safety and performance of treatment with a novel SAPD device in a small group of PD patients as a proof of concept.
Study objectives
The primary objectives are to assess (short-term) clinical and technical safety. Key secondary objectives include an evaluation of efficacy, patient tolerance, and uremic symptoms.
Study design
This is a first-in-human, prospective, open-label, non-randomized, single-arm, multicenter study (proof of concept), that will be performed at the University Medical Center Utrecht (UMCU; The Netherlands), Università degli studi di Modena e Reggio Emilia (UNIMORE, Italy), and Instituto de Investgacion Hospital Universitario La Paz, Servicio Madrileño de Salud (SERMAS, IdiPAZ, Spain). Inclusion of patients started in January 2024, and the study is expected to finish around June 2025.
Study population
The study population comprises adult PD patients who are stable on continuous ambulatory PD (CAPD) or automated PD (APD), have a well-functioning peritoneal catheter, and have no history of PD-related infection for at least 8 weeks prior to enrollment. In order to be eligible for study participation, a subject must meet all of the inclusion and none of the exclusion criteria (Table 1).
Inclusion and exclusion criteria.

PD: peritoneal dialysis.
Sample size
The pilot nature of this first-in-human study is descriptive and does not lend itself to a formal power analysis. We intend to enroll 12 subjects (6 treatments per subject) to assess (short-term) clinical safety and performance of the SAPD system in PD patients (proof of concept) since we expect that this sample size will provide reasonable first insight into safety and performance of the device and will capture sufficient (preliminary) information to adequately plan further steps of device development. Data of this first-in-human study will be used for sample size calculations in future studies.
Study endpoints
Primary endpoint
The primary safety objective will be assessed by describing and examining the incidence of:
Serious adverse device effects (SADEs) and device deficiencies (DDs) that could have led to a serious adverse event (SAE). Critical changes (requiring intervention) in patient's clinical condition and vital parameters (blood pressure, heart rate, body temperature, and oxygen saturation) during treatment. Critical changes (requiring intervention) in hematology and clinical chemistry (i.e. acid–base state, electrolytes) pre- vs post-treatment. Critical changes (requiring intervention) in intra-abdominal dialysate volume and intra-abdominal pressure.
Of note, “critical changes” were defined as any changes in the patient's condition or biochemical parameters which required intervention. To determine what a “critical” change was, we adhered to all local, national, or international clinical practices and—where necessary—clinical judgment. For instance, the dialysate effluent from the SAPD device was subject to specific requirements to guarantee patient safety (Supplemental Table 1).
Secondary endpoints
Incidence of adverse events and DD's other than SADE's and DD's that could have led to an SAE
Session characteristics (number, duration, dialysate flow rates)
(Evolution of) vital signs during treatment (blood pressure, heart rate, saturation, temperature)
Efficacy of UF in relation to glucose concentration in device effluent (UF volume/g absorbed glucose, maximum glucose concentration during treatment)
Efficacy of solute removal and base release of SAPD treatment
Absolute removal, MTAC, plasma clearance and plasma reduction ratio (RR) of urea, creatinine, phosphate, potassium, uric acid, beta-2 microglobulin, vitamin B12 and protein bound uremic toxins (PBUTs, only RR) Release of base (i.e. bicarbonate plus lactate)
Evolution of blood analytes (electrolytes, middle molecules, hematological parameters, liver function), measured before and after a dialysis session.
Evolution of device effluent parameters throughout a dialysis session
Device effluent analytes should be comparable to existing commercially available peritoneal dialysate formulations and should match the range of PD effluents during standard PD
To evaluate the (technical) device performance (DDs, adverse device effects, intraperitoneal pressure during SAPD treatment)
Patient tolerance (abdominal discomfort), rated on a numerical rating scale (NRS) at set timepoints throughout a dialysis session (at t = 0, 15, 60, 120, 180, 240, 480 min).
Evolution of uremic symptoms, scored using the dialysis symptoms index 18
Investigational device
The SAPD system comprises a wearable device with sorbents for use during the day (daytime system, 2.3 kg) that may be combined with a dialysate reservoir during the night (nighttime system, weight (filled) 12 kg). The 10L dialysate reservoir is filled with commercially available glucose-based dialysate, the glucose concentration being tailored to the patient's UF requirements. The nighttime system is intended to be used 7 days per week for 8 h per night to allow for sufficient solute removal and UF. Optionally, the wearable system can provide additional clearance of phosphate and non-urea organic waste solutes during the day (of note, in the current trial only the nighttime system will be tested). During SAPD treatment, only one peritoneal dialysate filling (Extraneal®, Baxter) per day is used. The system continuously recirculates 300 mL of peritoneal dialysate between the peritoneal cavity and the device by alternate in- and efflux via the patient's existing single-lumen peritoneal catheter (tidal flow; Figure 1), aiming for a mean effective dialysate flow rate of 75–100 mL/min. Flow rates can be reduced if the intended flow rate causes abdominal discomfort. Peritoneal effluent that enters the SAPD system is regenerated, which entails removal of uremic solutes and addition of glucose and bicarbonate (and/or lactate depending on dialysate used), after which it is returned into the peritoneal cavity (Figure 1). A new sorbent cartridge is used for each SAPD treatment session.
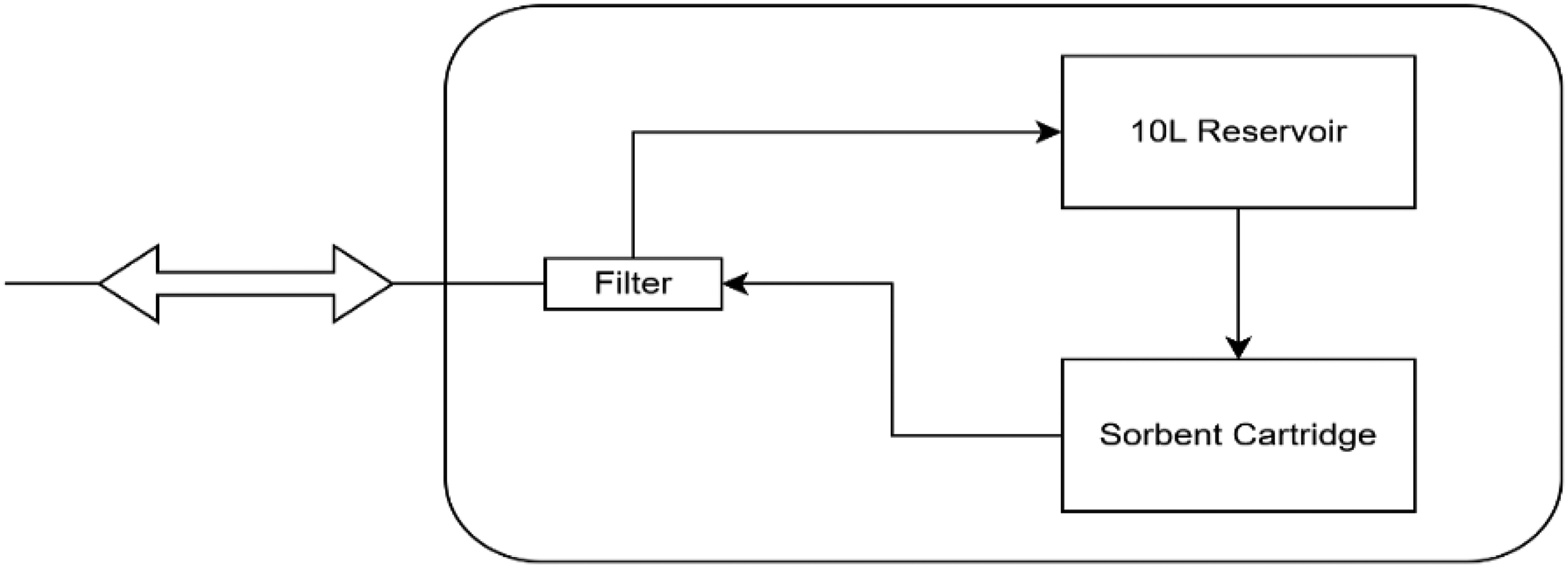
Schematic overview of the SAPD device. The patient is connected via the existing single-lumen peritoneal catheter, and alternating inflow and outflow of dialysate is applied by the device. Dialysate entering the device passes the filter and then enters the 10L reservoir. In the device OUT cycle, 300 mL of dialysate flows from the sorbent cartridge through the filter to the patient. At the same time, 300 mL is moved from the 10L reservoir to the fill up the sorbent cartridge again. SAPD; sorbent-assisted peritoneal dialysis.
The SAPD device was extensively tested in vitro 19 and in vivo. 20 In short, in vivo efficacy of the SAPD device was higher compared to conventional dialysis, and no safety concerns were observed. The treatment was apparently well tolerated by the animals without any observed discomfort, adverse reactions, or abnormal lab values (a.o. electrolytes, acid–base balance, hematological parameters). In addition to these tests, a comprehensive biological evaluation was performed according to the ISO-10993 guidelines, in which no toxicological or biological risks were identified. Additionally, a formative usability trial in nurses and patients was performed to identify any design issues that may impair successful use of the device. The SAPD system was found to have a slightly higher than average usability for its design stage (measured using the System Usability Score), and useful insights to prevent potential critical use error and general feedback to prevent critical use errors were obtained. This input was used to make further improvements to the device ahead of the clinical testing phase.
Study procedures
Informed consent
Nephrologists at the participating centers will ask eligible patients whether they may be approached by the study team. The study team will then give/send the patient information and—if the patient is interested in participating—plan a visit to sign the informed consent form. During this visit, the patient can ask any questions, after which informed consent is signed. This is followed by a short physical examination (lung auscultation, presence of peripheral edema, vital signs). Additionally, demographical data, medical history, current medication use, and current PD prescription is collected from the participant's file and will be verified with the participant. Based on this, comorbidity is scored using the Charlson Comorbidity Index.21,22 Materials for the 24 h collections (see below) will be provided at the conclusion of this visit. Finally, a serum pregnancy test is performed in all female patients ≤55 years of age.
Baseline
After written informed consent has been obtained, the patient will be asked to perform 24 h collections of both urine and dialysate on 3 (consecutive) days. An overview of all measured solutes is displayed in Supplemental Table 2–9. Presence and degree of abdominal discomfort will be assessed during each baseline visit using the NRS. A blood sample will also be drawn in the hospital on each of these days. The third day a peritoneal equilibration test (PET) 23 will be performed in the hospital. To be able to assess MTAC more accurately during the PET, we opted to use a Physioneal 1.36% (Baxter) solution (as opposed to a high e.g. 3.86% solution), in order to limit UF and thus convective solute transport.
Sorbent-assisted peritoneal dialysis treatment days
SAPD treatment is scheduled in the 2 weeks directly after the baseline measurements. The SAPD system is tested in a clinical setting for a total of 6 days spread over 2 weeks (Figure 2). All treatments will be performed during daytime in the hospital to allow continuous monitoring of the participants.
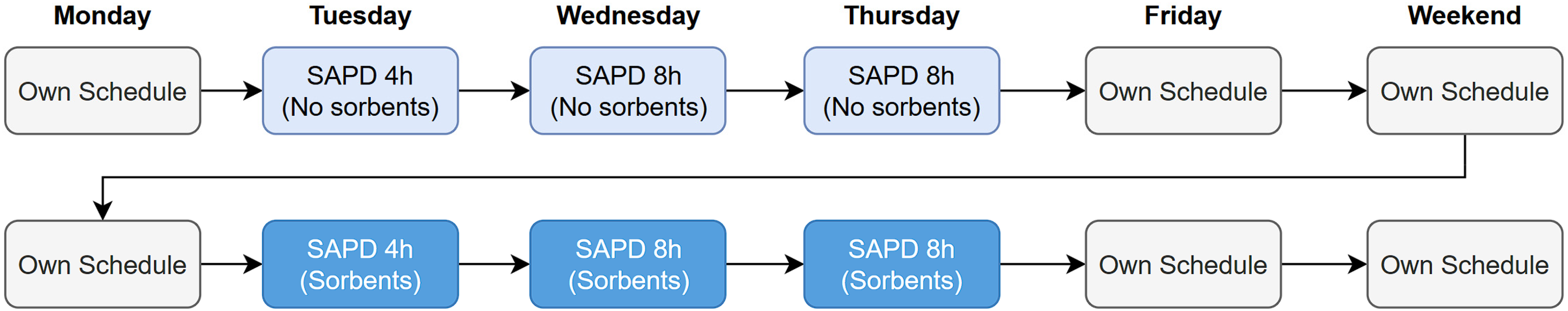
Overview of SAPD treatment procedure per individual patient—of note, all SAPD treatments will be performed during daytime in the hospital. Overnight, in between the SAPD treatments, patients will receive an Extraneal® dwell. After the last SAPD treatment, patients will resume their regular PD schedule. SAPD: sorbent-assisted peritoneal dialysis; PD: peritoneal dialysis.
During the first week, patients will be treated with the SAPD nighttime system without sorbents for 4 h (1st treatment day) and 8 h (2nd and 3rd treatment day). The night before the first treatment and in between treatment days, patients will receive 1L Extraneal® instead of their regular PD schedule. With this, filling patients will come to the hospital, where a sample is drawn from the overnight dwell and a partial fill (up to an intraperitoneal volume of ∼1.5 L) is performed in order to accurately measure the intraperitoneal volume at the start of the SAPD treatment. This is required for accurate measurements and calculations of UF volume, MTAC, and clearances. Of note, this partial fill procedure will not be part of the final intended use procedures. At the end of the treatment, an exchange with an Extraneal® solution is performed and residual volume is determined based on total albumin concentration in the peritoneal dialysate in the complete drain after SAPD treatment, and after installation of the fresh Extraneal®. It should be noted that the intraperitoneal icodextrin solution present at initiation of SAPD treatment becomes progressively diluted by the glucose-based dialysate in the reservoir during the course of the treatment. Finally, after receiving the last treatment of the week, patients on CAPD will continue their regular glucose-based schedule, whereas APD patients receive a fresh Extraneal® filling and resume their APD schedule the same or following day, at the physician's discretion.
Treatment during the second week is similar to the first week in all regards but the fact that the SAPD nighttime system with sorbents is used. This strategy, together with the first treatment of the week being 4 instead of 8 h, provides incremental exposure to the two novel elements of SAPD treatment, i.e. 1) continuous flow of peritoneal dialysate, and 2) sorbent technology. In this way, we can separately evaluate the effect of the continuous flow and use of sorbents.
Follow-up
The week after the second week with SAPD treatment, the patient will return to the hospital for another PET, after which study follow-up is completed.
All patients will be treated with the SAPD system consecutively, enabling thorough analysis of the data (efficacy, safety, technical performance, tolerance) by the research team and—if required—an independent data safety monitoring board (DSMB) before the next patient receives SAPD treatment. This way, the number of subjects simultaneously put at risk will be minimized.
Study measurements
During SAPD treatment, the patient's clinical condition is closely monitored in a clinical setting. Blood pressure, heart rate, respiratory rate, and body temperature are measured at regular intervals during treatment. Body weight is measured before and after each SAPD treatment. Patients are encouraged to report all possible adverse events. Abdominal (dis)comfort is scored at baseline during three conventional filling and drain procedures and during a dwell (nine times in total) using a numeric rating scale (0–10). During SAPD treatment, abdominal discomfort is scored before start and regularly during treatment (after 15, 60, 120, 240, and 480 min), and more frequently if the patient experiences abdominal discomfort or pain.
Sampling is performed continuously during a tidal flow cycle, since solute concentrations are not stable during the in- and outgoing phase as a result of the dead volume of the filter and peritoneal catheter. The dead volume in the sampling line is discarded before each sample is taken. The dialysate sampling points are located between the filter and the rest of the SAPD system, and not at the side of the patient's peritoneal catheter to prevent microbial contamination. Of note, during the first-in-human clinical trial, all (dis)connections of the peritoneal catheter and sampling procedures will be performed under strict hygienic operating procedures by trained study personnel to prevent user errors.
A complete assessment schedule and overview of all laboratory parameters measured for this study are detailed in Supplementary tables 2–8.
Data analysis
Demographics, baseline characteristics, and safety variables will be summarized by descriptive statistics; continuous variables will be reported as mean (standard deviation) or median (interquartile range) as appropriate. Categorical variables will be reported as counts and/or number of cases (%). For categorical data, a chi-square analysis will be performed. The difference between baseline and treatment parameters and the change between pre- and post-treatment parameters, including laboratory measurements, clinical condition of the patient, and abdominal discomfort, will be analyzed using a paired t-test or Wilcoxon signed rank test as appropriate. If the data distribution allows, mixed models or repeated-measure analyses may also be used. Treatment effects will be considered statistically significant at p < 0.05 (two-tailed). In case of missing data, no imputation will be performed. Of note, besides the interim assessments for treatment safety to decide on continuation of treatment, no formal (statistical) interim analyses will be performed. All statistical analysis will be performed using SPSS, SAS, R, STATA, Graphpad Prism, or other widely accepted statistical software.
Ethical considerations
This first-in-human clinical trial will be conducted in accordance with the principles of the Declaration of Helsinki (64th amendment, Brazil, October 2013). In addition, the study will be performed in compliance with relevant international and local legislation/guidelines such as the MDR (article 62), data protection regulation (GDPR), and good clinical practice (ISO 14115). Ethical approval has been obtained in The Netherlands (NedMec, 23-075/H-A) and in Italy (EC AVEN ProtAOU 0014371/24 15May2024, MoH 0049307-11/06/2024-DGDMF-MDS-P); efforts are currently underway to obtain approval in Spain. The study has been registered in clinicaltrials.gov (NCT06314503).
Data safety monitoring board
An independent DSMB is installed to monitor the safety and overall conduct of the clinical trial according to present best practice as described in the DAMOCLES study. 24 The DSMB consists of a nephrologist, an epidemiologist, and a statistician. The study team will report all relevant information on safety and study progress to the DSMB. All events, including death, will be formally evaluated by the DSMB at predetermined milestones: after the treatment of the first two patients, and after finishing treatment of all patients for a respective participating center. Additional meetings can be called if the study safety officer determined a necessity thereto.
Discussion and conclusion
Here, we present the protocol for a comprehensive first-in-human study of a novel SAPD device. To our knowledge, this is the first study to separately assess the effect of the different technical aspects (i.e. the continuous flow and the added value of use of sorbents). Furthermore, a broad set of outcomes and efficacy parameters is measured to meticulously assess the technical and clinical performance of the device, as well as the patient tolerance and safety. Taken together, this study may yield results that have broader relevance for similar initiatives in the field.
Inclusion and treatment of patients is currently ongoing and database lock is expected to take place around June 2025. Preliminary results are being evaluated to identify potential improvements and to inform the design of future clinical studies which should ultimately pave the way for CE-marking ahead of market introduction.
Supplemental Material
sj-docx-1-ptd-10.1177_08968608251381933 - Supplemental material for Rationale and design of the CORDIAL first-in-human clinical trial: A system for sorbent-assisted continuous flow peritoneal dialysis
Supplemental material, sj-docx-1-ptd-10.1177_08968608251381933 for Rationale and design of the CORDIAL first-in-human clinical trial: A system for sorbent-assisted continuous flow peritoneal dialysis by Joost Christiaan de Vries, Maaike K van Gelder, Dian P Bolhuis, Frank Simonis, Marianne C Verhaar, María Auxiliadora Bajo Rubio, Gloria del Peso, Rafael Selgas, Gabriele Donati, Giulia Ligabue, Gianni Cappelli and Karin GF Gerritsen in Peritoneal Dialysis International
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: CORDIAL is an investigator-driven trial. The project received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No. 733169 and No. 945207, and by the Dutch Kidney Foundation and Dutch Ministry of Economic Affairs by means of a PPP Allowance made available by the Top Sector Life Sciences & Health to stimulate public private partnerships (DKF project code PPS08).
Author contributions
All authors were involved in study design; JdV, MvG, and KG wrote the first draft; All authors reviewed and edited the final draft.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.