
Abstract
This study systematically examined the effects of functional groups (-O, -OH, and -COOH), temperature (200 K-500 K), and defects (single vacancies, double vacancies, and topological defects) on the interfacial structure and mechanical properties of graphene (Gr)/graphene oxide (GO) composites with hydrated calcium silicate (C-S-H). Experiments show that adding Gr and GO significantly increases the compressive and flexural strength of the composite, with GO exhibiting a stronger strengthening effect. Simulation results indicate that oxygen-containing functional groups are key to enhancing interfacial bonding, with the performance improvement attributed to the coordination and hydrogen bonding interactions between polar functional groups and Ca2+.Temperature strongly influences material properties. When the temperature rises from 200K to 500K, the peak values of the radial distribution functions for Ca-Ow and Ca-Oc decrease significantly. At the same time, the rate of carbon atom migration increases sharply, leading to a decline in the mechanical properties of the composite. However, the presence of hydroxyl and carboxyl groups can effectively reduce this high-temperature degradation. Defects cause minor fluctuations in the peak values of the radial distribution functions for Ca-Ow and Ca-Oc. Under tensile and compressive loads, defects lead to small changes in mechanical properties, with some composites showing slight improvements. Overall, defects have a much weaker impact on the interfacial structure and mechanical properties compared to functional groups and temperature effects. This research clarifies the key mechanisms underlying the effects of functional group modification, temperature regulation, and defects, offering essential theoretical insights to improve the mechanical properties and high-temperature stability of cement-based composites.
Introduction
Cementitious materials, among the most commonly used in construction engineering,1–3 directly influence structural safety and service life through their mechanical properties and durability.4,5 However, the inherent brittleness and poor crack resistance of traditional cementitious materials make them vulnerable to progressive damage under complex service conditions, such as temperature fluctuations and mechanical impacts, making it difficult to meet modern engineering demands for high strength, toughness, and durability.6–8 Therefore, developing effective methods to optimize the microstructure and mechanical properties of cementitious materials through scientifically based modification techniques has become a crucial challenge in civil engineering and materials science.
Calcium silicate hydrate (C-S-H), the main product of cement hydration reactions,9,10 accounts for over 60% of the cement volume. The density of the C-S-H microstructure and the quality of interfacial bonding directly influence the mechanical properties and service life of cementitious materials, serving as the core mechanism for controlling material performance.11,12 In recent years, nano-reinforcement technology has offered a breakthrough approach for modifying cement-based materials. Among these, graphene (Gr) and graphene oxide (GO) are seen as highly promising reinforcing materials due to their extremely high specific surface area, excellent mechanical properties, and unique chemical reactivity.13–15 Gr features a perfectly sp2-hybridized six-membered carbon ring and, in theory, exhibits very high tensile strength. However, its surface is inert and hydrophobic, leading to poor compatibility with the cement matrix and a tendency to agglomerate, which greatly limits its reinforcing potential.16,17 As a derivative of Gr, GO is rich in oxygen-containing functional groups such as hydroxyl (-OH), carboxyl (-COOH), and epoxy (-O) groups on its surface. These groups can form chemical and hydrogen bonds with cement hydration products, strengthening interfacial interactions and improving material performance.18–20 Previous studies have systematically examined the adhesion stability of -COOH, -OH, and -O functional groups on the edges and surfaces of graphene nanoribbons, revealing how functionalization modifies the electronic structure and quantum transport properties of graphene, 21 laying a theoretical foundation for designing functional group configurations in graphene-based materials. Meanwhile, covalent bonding between GO and polyacrylamide significantly enhances the composite’s mechanical properties. This modification method offers valuable insights for designing interface reinforcement between GO and cement hydration products. 22 Changes in size and chemical functionalization can also modulate the optical bandgap of graphene-based materials, confirming the vital role of functional group modification in controlling the electronic structure and band properties of graphene, 23 and further refining the theoretical framework for GO functionalization. Reddy et al. 24 observed that adding small amounts of GO to concrete significantly enhances mechanical properties while reducing workability. Incorporating GO into concrete promotes hydration reactions and helps form a uniform, dense microstructure. Li et al. 25 studied the early-stage microstructure of graphene-cement composites via scanning electron microscopy (SEM). SEM images showed that needle-like sulfoaluminates, C-S-H gel, and other hydration crystals were interconnected by graphene flakes, creating a three-dimensional structure capable of bridging cracks and filling pores within the cement matrix. Chen et al. 26 demonstrated that adding 0.02 wt% GO and GNP can increase flexural strength by 16.3% and 11.6%, respectively. Moreover, GO and GNPs can fill cracks and help form a dense microstructure within the cement mortar matrix.
Although experimental studies have confirmed that Gr and GO strengthen cementitious materials, the mechanisms of their interfacial interactions with the C-S-H matrix remain unclear. Wang et al. 27 observed that the interfacial bonding strength between GO and C-S-H was significantly higher compared to that between Gr and C-S-H, due to the stability of chemical bonds and mechanical interlocking. The interfacial bonding strength decreased with lower moisture content, indicating that infiltrating water weakens the adhesion between GO and C-S-H. Existing research has mainly focused on the effects of individual factors on the properties of composite materials. However, in practical engineering situations, the in-service performance of materials is often affected by multiple factors. The strengthening mechanisms benefit from the presence of various oxygen-containing functional groups on the GO surface, which promote interfacial bonding. Inevitable defects during material preparation and service, such as single vacancies, double vacancies, and topological defects, impact stress transfer and failure behavior in composite materials. Additionally, temperature variations can influence the bonding properties of graphene-based materials in cementitious matrices, thereby affecting the mechanical properties of the composite structure. The chosen temperature range of 200-500 K aims to cover the spectrum from the low-temperature brittle zone to thermal aging or high-temperature curing conditions that cement-based materials may experience, systematically revealing how interfacial properties evolve with temperature. These key scientific questions have yet to be thoroughly addressed, which greatly limits the precise design and application of high-performance nano-modified cement-based materials.
Molecular dynamics (MD) simulations offer a visual portrayal of how material microstructures evolve and reveal the mechanisms behind interfacial interactions, providing theoretical support for macroscopic experimental data. By analyzing key parameters such as the radial distribution function, mean-squared displacement, and stress-strain curves at the molecular level, these simulations help clarify how various factors influence the tensile, compressive, and Young’s moduli of composite materials. This study aims to uncover the fundamental nature of the interface interaction between Gr (GO) and the C-S-H matrix. It establishes theoretical foundations and technical guidance for enhancing the mechanical properties and high-temperature stability of cementitious composites through functional group modification, defect regulation, and temperature adaptation. Ultimately, this research promotes the broader application of nano-modified cementitious materials in practical engineering.
Modeling and simulation methods
The construction of the calcium silicate hydrate (C-S-H) model is based on Pellenq’s
28
modeling approach. The initial structure uses the 4a×3b × c supercell of Tobermorite 11 Å (Ca/Si = 1). All water molecules and OH groups are removed from the structure, then the monoclinic supercell is converted to an orthorhombic supercell. Next, SiO2 groups on the silicon chains within the supercell are randomly removed to match the Qn distribution. In this study, the Qn distribution is Q0 = 4.7%, Q1 = 74.4%, and Q2 = 20.9%, which closely matches the results obtained by NMR.
28
Energy minimization and structural relaxation are then performed using the Conjugate Gradient (CG) method. Water molecules are adsorbed using the Giant Conjugate Monte Carlo (GCMC) method at 300 K and a chemical potential of 0.5 eV. The adsorbed structures are relaxed for 100 ps under the NPT ensemble. The final chemical formula for the C-S-H structure is (CaO)1.67(SiO2)1.8(H2O), with a crystal cell density of 2.4 g/cm3. The water-absorbed equilibrium model is then replicated 2 × 2 × 4 times along the X, Y, and Z directions. A 10 Å nano-crack is created along the Z direction (the weakest direction) in the C-S-H model. Gr and GO are introduced into this nano-crack to form the composite material, as shown in Figure 1. Cracked C-S-H model and molecular structure with Gr insertion.
The graphene model is based on a single-crystal graphite crystal with unit cell parameters of 2.46 Å × 4.26 Å × 3.4 Å. The fundamental unit cell is expanded by a factor of 17 in the X direction and 21 in the Y direction to create the final graphene structure. Experimental studies indicate that the proportion of oxygen-containing functional groups relative to the number of carbon atoms in GO is set at 20-30%.
29
The oxygen-containing functional group coverage R in the GO disordered structure model is defined as the ratio of the functional group count to the number of carbon atoms in graphene. As shown in Figure 2, different types of oxygen-containing functional groups were randomly distributed on both sides of the graphene nanosheets, including 20% epoxy groups (-O), hydroxyl groups (-OH), and carboxyl groups (-COOH). These include graphene (Gr), epoxy-functionalized graphene oxide (GO-O), hydroxyl-functionalized graphene oxide (GO-OH), and carboxyl-functionalized graphene oxide (GO-COOH). To clearly explain the performance characteristics and functional mechanisms of each composite model, the functional group-modified composites are designated as Gr/C-S-H, GO-O/C-S-H, GO-OH/C-S-H, and GO-COOH/C-S-H, respectively. Gr, GO-O, GO-OH, and GO-COOH models.
For the atomic interaction characteristics of different components, the C-S-H phase uses the ClayFF force field,30,31 which assigns interatomic forces to non-bonded interactions. This force field has been successfully applied to study the mechanical and elastic properties of clay-based nanocomposites and clay-based minerals.32,33 In Gr and GO, atomic interactions are simulated using the CVFF force field. The CVFF force field accounts for bond lengths, bond angles, torsion angles, and dihedral angles, and it has been validated for accurately simulating the structure and mechanical properties of graphene and its derivatives.34–37 For short-range van der Waals dispersive interactions, the Leonard-Jones (12-6) potential is used, with calculations following the geometric guidelines of the combined ClayFF and CVFF force fields. Studies have shown that the combined use of ClayFF and CVFF can accurately characterize the interaction mechanisms in inorganic-organic systems, supporting the validity of these force fields.38–42 Relevant parameters are listed in Reference 43. The study separately examined the effects of temperature and defects. Tensile and compressive simulations of the Gr (GO)/C-S-H composite were conducted at specific temperatures of 200 K, 300 K, 400 K, and 500 K. Different defect models were simulated at 300 K. The simulations used a time step of 0.1 fs, consistent with prior studies.44,45 The long-range Coulomb potential was calculated using the Ewald summation method. The simulation procedure was as follows: first, structural optimization was performed with a convergence criterion of 10−8 kcal/mol. Then, pressures in the X, Y, and Z directions were set to zero. The Gr (GO)/C-S-H composite was placed in an NPT ensemble between 200 K and 500 K and relaxed for 100 ps. For composites with different defect types, relaxation at 300 K for 100 ps was conducted to reach equilibrium at the target temperature. To enable full interaction between Gr (GO) and C-S-H, the system was operated in an NVT ensemble at the target temperature for 100 ps, followed by an additional 300 ps to generate atomic trajectories for analysis. Tensile and compressive simulations were performed at a constant strain rate of 0.008/ps, within the typical range used in molecular dynamics simulations of cement-based composites (1 × 10−2/ps to 1 × 10−4/ps). 46 Periodic boundary conditions were applied throughout the simulation to reduce boundary effects.
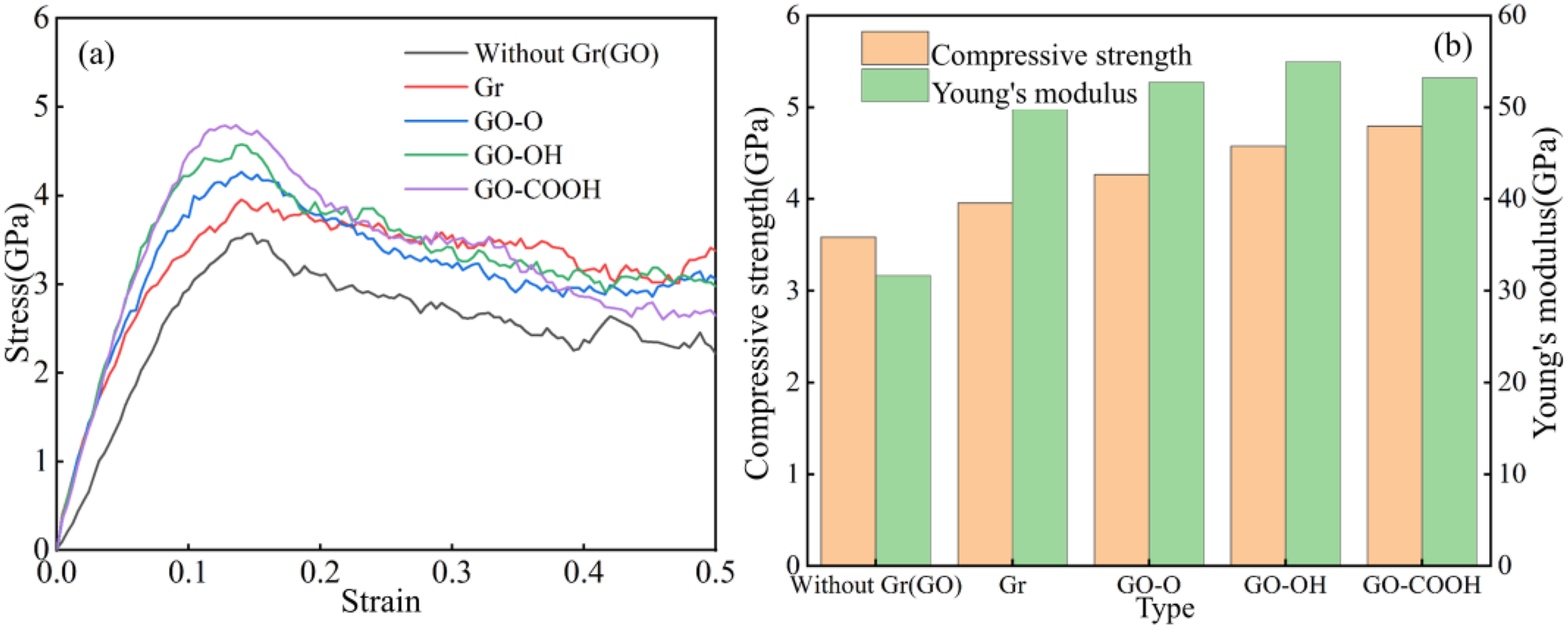
Before conducting molecular dynamics (MD) simulations, the compressive and flexural strengths of ordinary cement mortar (PC), cement mortars modified with different amounts of graphene (Gr), and cement mortars modified with various amounts of graphene oxide (GO) were first tested using a universal testing machine, as shown in Figure 3(a)–(d). The baseline compressive strength of PC is 42.57 MPa, and its flexural strength is 6.76 MPa. The compressive strength of Gr-modified specimens initially increases and then decreases with increasing dosage, reaching a peak of 45.39 MPa at 0.075% dosage, about 6.6% higher than that of the PC group. Increasing the dosage to 0.12% further reduced the compressive strength to 44.06 MPa, but it remained above the baseline. Similarly, flexural strength peaked at 8.88 MPa at 0.075% content, showing an approximately 31.4% increase over the PC group. At 0.12% content, flexural strength was 8.69 MPa, still relatively high. In contrast, GO-modified specimens demonstrated more significant reinforcement effects. Compressive strength reached a maximum of 48.65 MPa at 0.03% GO content, representing an approximately 14.3% increase over the PC group. When GO content was increased to 0.05%, compressive strength fell to 45.07 MPa. Flexural strength peaked at 9.08 MPa with 0.03% GO, about 34.3% higher than that of the PC group. This value also surpassed the flexural strength of the Gr-modified specimens at the same filler content. Further increasing to 0.05% GO reduced flexural strength to 7.69 MPa, but it remained higher than that of the PC group. Compressive strength and flexural strength of cement-based composites with graphene and graphene oxide as additives, varying with the content of graphene and graphene oxide.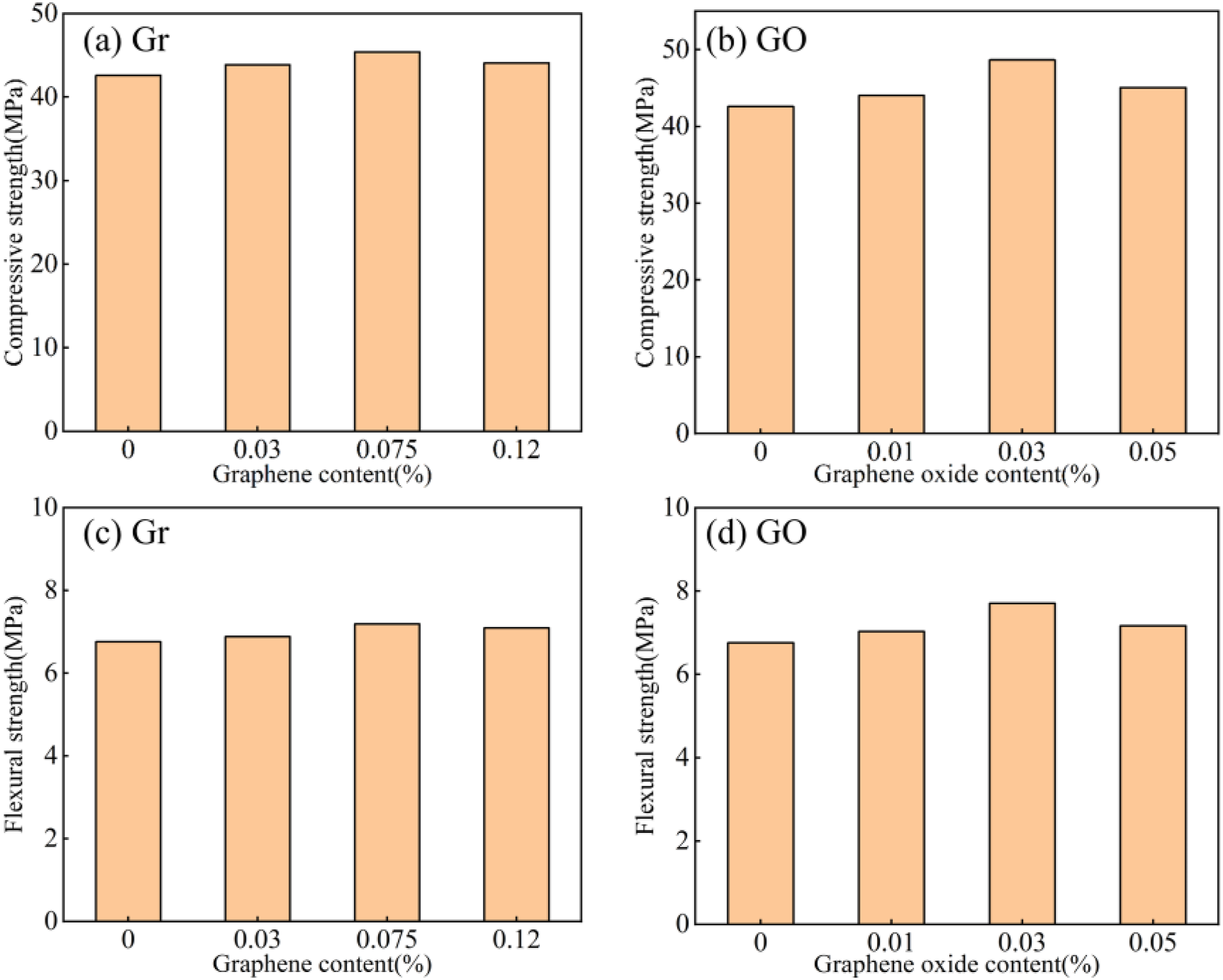
Overall, the appropriate amounts of graphite and graphene oxide effectively improve the compressive and flexural strengths of cement mortar. Notably, the gain in flexural strength, which is closely linked to toughness, surpasses that of compressive strength by a significant margin. This highlights the important role of nanomaterials in reducing the brittleness of cement mortar. At the optimal dosage, GO-modified specimens exhibit significantly higher compressive and flexural strengths than Gr-modified specimens, indicating that GO provides greater enhancement of the cement mortar’s mechanical properties. This is mainly due to the abundant oxygen-containing groups on the GO surface, which form stronger interfacial bonds and chemical links with cement hydration products (such as calcium hydroxide and calcium silicate hydrate). This promotes efficient stress transfer and prevents microcrack growth. The hydrophobic surface of Gr is poorly compatible with the cement matrix, leading to particle clumping and weakening its reinforcement. When the dosage exceeds the optimal level, both nanomaterials show a decline in strength. This suggests that a high dosage leads to uneven nanoparticle dispersion within the matrix, creating stress-concentration zones that disrupt the microstructure and reduce mechanical properties. The results on mechanical properties offer crucial macroscopic performance evidence for later MD simulations to clarify how nanomaterials interact with the cement matrix.
Results and discussion
Interface structure and mechanical properties of Gr (GO) and C-S-H composites
After forming composites with Gr and GO containing different functional groups and C-S-H, the radial distribution functions (RDF) of Ca-Ow, Ca-Os, and Ca-Oc in the composites were calculated. Among these, Ca represents interlayer calcium ions in C-S-H, Os denotes oxygen atoms in silicate tetrahedra within C-S-H, Oc signifies oxygen atoms on the surface of GO, and Ow refers to oxygen atoms in water. As shown by the RDF plots in Figure 4(a)–(d), the introduction of Gr/GO significantly alters the local coordination environment of Ca2+ in C-S-H. In all systems, the Ca-Os bond exhibits the strongest characteristic peak at 2.29 Å, confirming the dominant influence of the siloxane framework on Ca2+. In contrast, the intensity of the Ca-Ow peak at 2.44 Å is greatly reduced. Notably, the GO-OH/C-S-H and GO-COOH/C-S-H composite systems show higher Ca-Ow peak intensities because hydroxyl (-OH) and carboxyl (-COOH) groups have strong polarity and coordination ability. Their surface oxygen atoms (Oc) form stable bonds with Ca2+ and also anchor additional water molecules through hydrogen bonds, strengthening the interaction between water and Ca2+. This mechanism demonstrates how functional groups indirectly affect interfacial bonding strength by modifying the structure of interfacial water. Since the surface of Gr/C-S-H lacks oxygen-containing functional groups, its Ca-Ow peak is lower than in GO-modified systems, emphasizing the key role of oxygen-containing groups in regulating C-S-H. The RDF curves for Ca-Oc show that the characteristic interaction distances for GO-O/C-S-H, GO-OH/C-S-H, and GO-COOH/C-S-H are 2.51 Å, 2.48 Å, and 2.44 Å, respectively. The decreasing coordination distances with increasing functional group polarity directly indicate that the binding between Ca2+ and surface oxygen (Oc) on GO becomes stronger. Figure 5 presents the mean squared displacement (MSD) curves for carbon atoms in the Gr (GO)/C-S-H composites. The MSD for Gr/C-S-H is the highest and increases rapidly, indicating that carbon atoms in Gr are the most mobile. This is because only weak van der Waals forces exist between the inert Gr surface and the C-S-H group, providing limited interfacial confinement. When oxygen-containing functional groups are added, the mobility decreases in the order of the systems, with GO-OH and GO-COOH showing the lowest MSD values. This suggests that hydroxyl and carboxyl groups significantly enhance interfacial constraints by coordinating with Ca2+ and forming hydrogen bonds, thereby effectively suppressing the thermal migration of carbon atoms and improving interfacial structural stability. RDF of Gr (GO)/C-S-H composites: (a) Gr/C-S-H; (b) GO-O/C-S-H; (c) GO-OH/C-S-H; (d) GO-COOH/C-S-H. MSD curve of carbon atoms in Gr (GO)/C-S-H composite materials.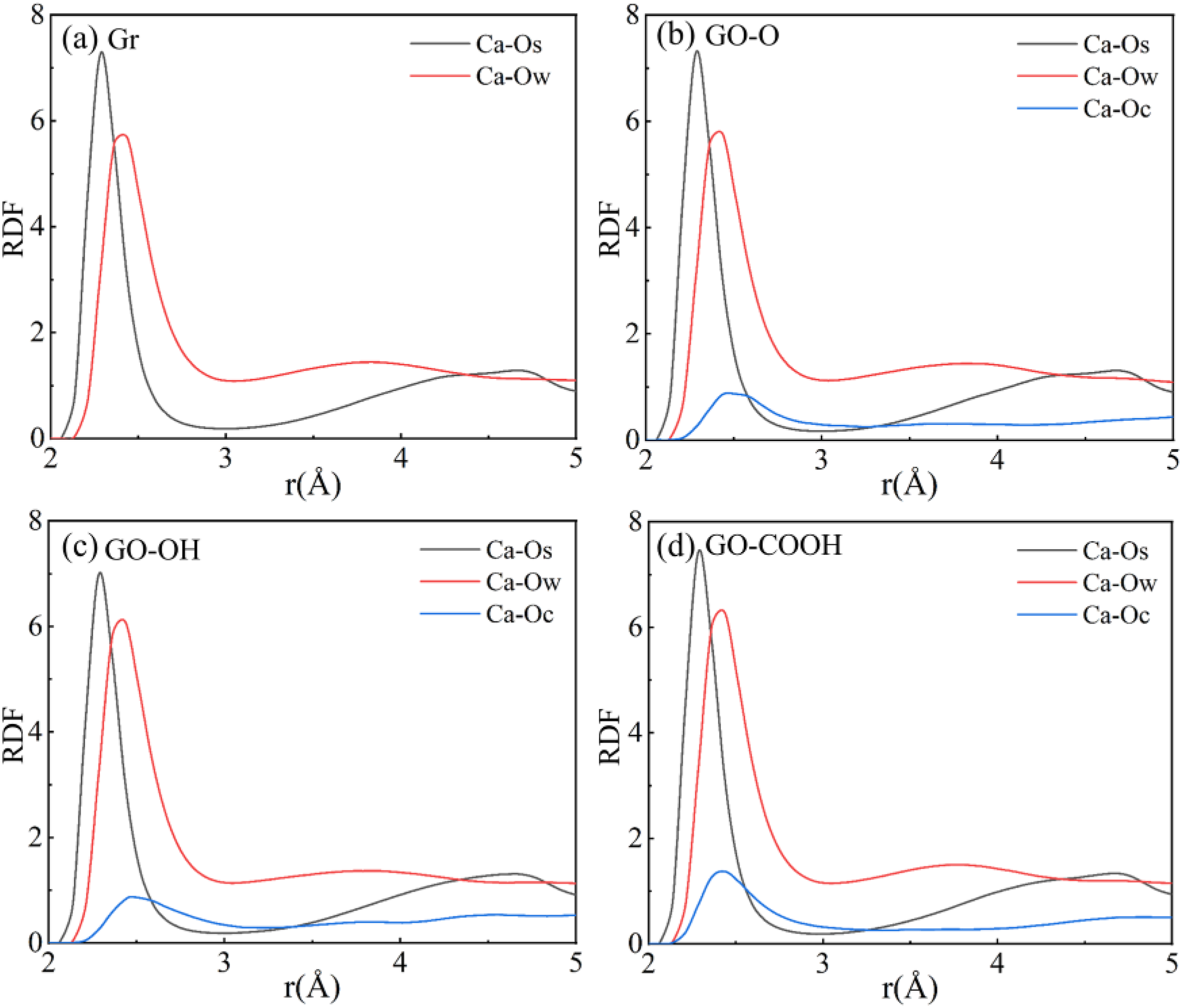
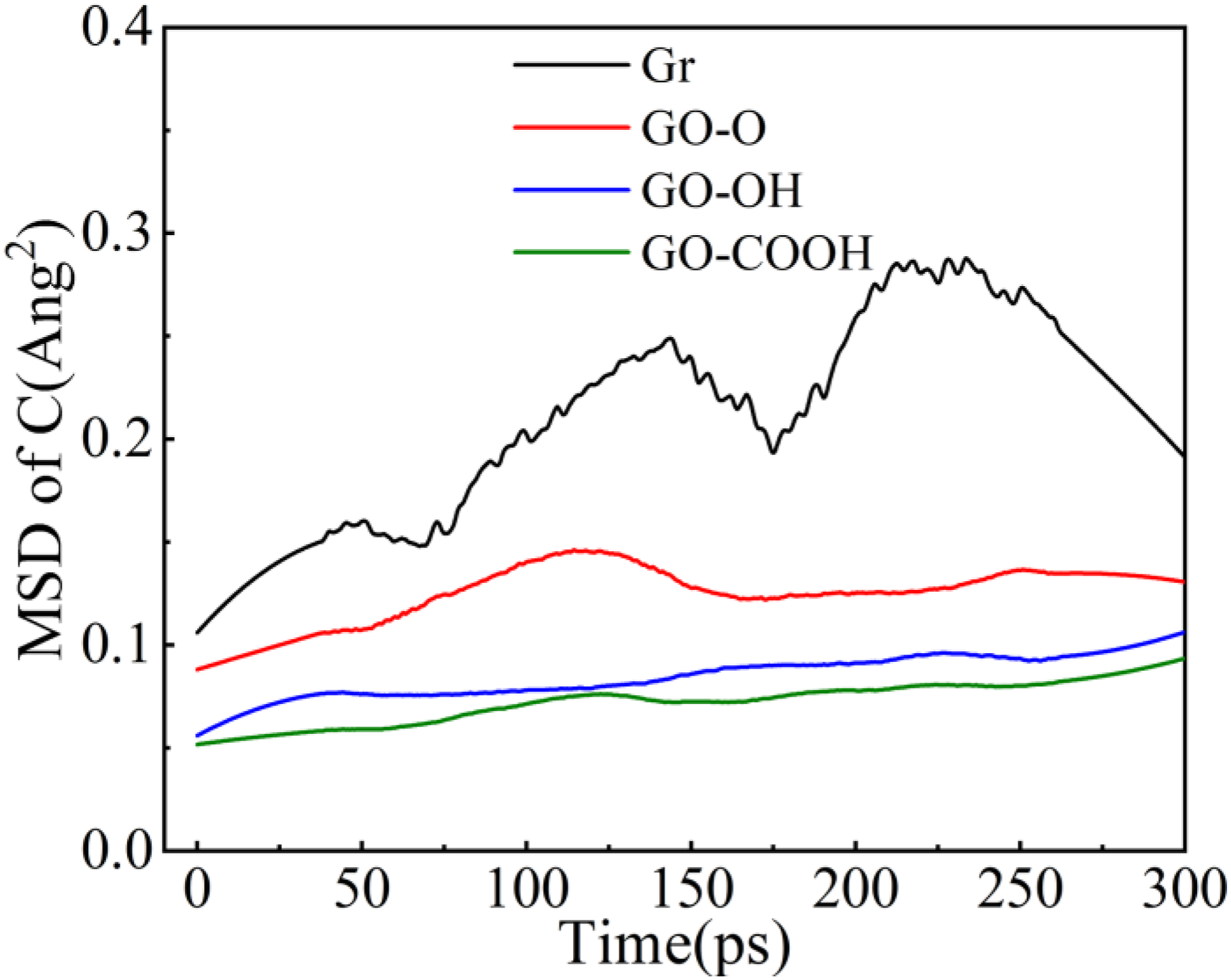
Figure 6(a)–(b) show the stress-strain curves, tensile strength, and Young’s modulus of Gr (GO)/C-S-H under tensile load along the z-direction. As shown in Figure 6(a), C-S-H has the lowest peak tensile stress. With the addition of Gr and GO, interfacial bonding and load transfer improve, leading to a significant increase in both peak stress and fracture strain, with GO-COOH/C-S-H displaying the highest load-bearing capacity. This trend aligns with the ranking of interfacial coordination strength and functional group activity, highlighting the important role of interfacial interactions in tensile properties. Figure 6(b) features a bar chart comparing the tensile strength (orange bars) and Young’s modulus (green bars) of different composites. The tensile strength of C-S-H is 1.53 GPa, closely matching the 1.51 GPa reported by Lin et al..
47
The elastic modulus is 23.53 GPa, within the experimentally observed range of 20-30 GPa.48,49 The addition of Gr and GO significantly boosts both the tensile strength and Young’s modulus of the composite. The failure model in Figure 7 further confirms these changes in mechanical properties. At a strain of 0.4, the greatest extent of interfacial cracking occurred at the C-S-H interface. Cracks at the Gr/C-S-H interface became more concentrated along the z-direction. The GO/C-S-H interface showed less damage, with microcrack propagation being suppressed. The GO-OH/C-S-H and GO-COOH/C-S-H interfaces exhibited the strongest bonding, with only minor localized damage. As shown in Figure 8(a)–(b), the compressive strength (3.58 GPa) and Young’s modulus (31.65 GPa) of C-S-H are relatively low, while the introduction of Gr (GO) effectively enhances the material’s compressive properties. Gr (GO)/C-S-H stretched along the z-direction: (a) stress-strain curve; (b) tensile strength and Young’s modulus. Failure models of C-S-H and Gr (GO)/C-S-H composites under tensile loading along the z-direction at a strain of 0.4. Gr (GO)/C-S-H compressed along the z-direction: (a) stress-strain curve; (b) compressive strength and Young’s modulus.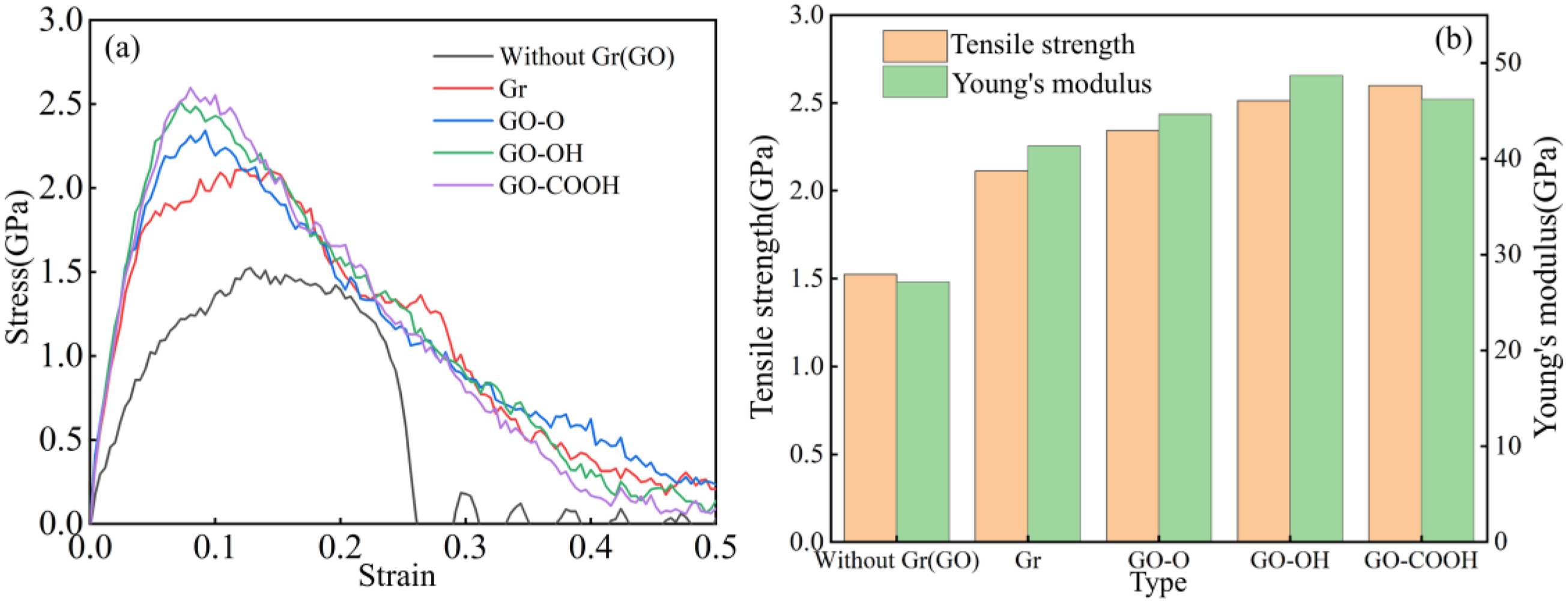

Effect of different defect types on the interface structure and mechanical properties of Gr/GO and C-S-H composites
Three defect types were created on the surfaces of Gr and GO: single vacancy (SV), double vacancy (DV), and a topological (SW) defect featuring a 90° rotation of the carbon-carbon bond. For SV and DV defects, the defect concentration refers to the proportion of missing carbon atoms relative to the total number of carbon atoms in pristine graphene. For SW defects, the defect concentration generally refers to the proportion of rotated carbon atoms relative to the total number of carbon atoms in pristine graphene. Previous studies have shown that concentrations between 2% and 5% fall within the low-defect range and have minimal impact on the mechanical properties of composite materials. Performance deteriorates significantly only when the concentration reaches 10%. Therefore, this paper selects 2% as the defect concentration, enabling a fair comparison of the behavior patterns of different defect types without damaging the graphene structure. Figure 9(a) shows graphene (Gr-SV/DV/SW) with various defect types, while Figure 9(b) depicts graphene oxide containing epoxy groups (GO-O-SV/DV/SW) under different defect configurations. Figure 9(c) shows graphene with hydroxyl groups under various defect types (GO-OH-SV/DV/SW), and Figure 9(d) depicts graphene with carboxyl groups under different defect types (GO-COOH-SV/DV/SW). (a)–(d) shows the Gr, GO-O, GO-OH, and GO-COOH models for different SV, DV, and SW defects.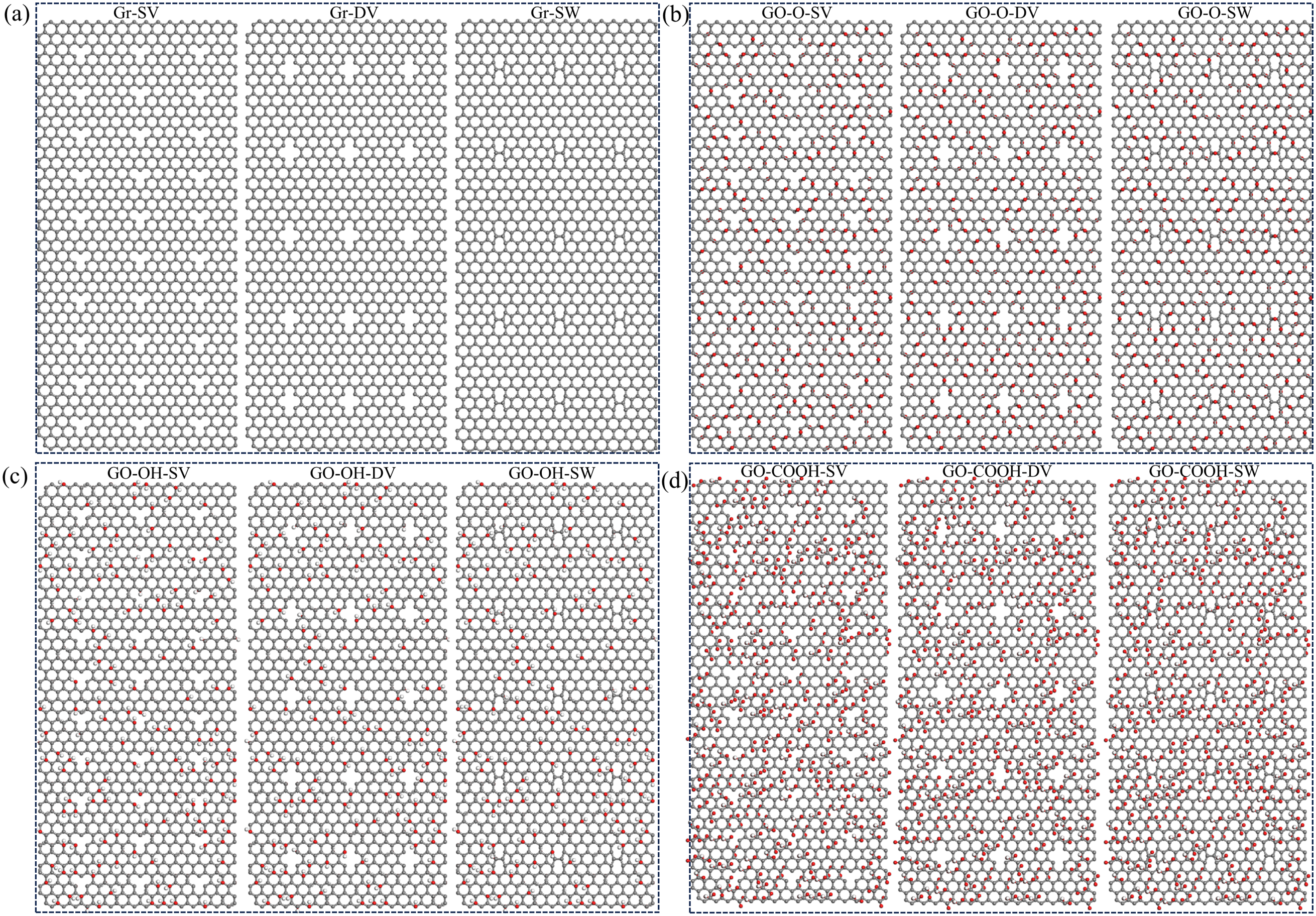
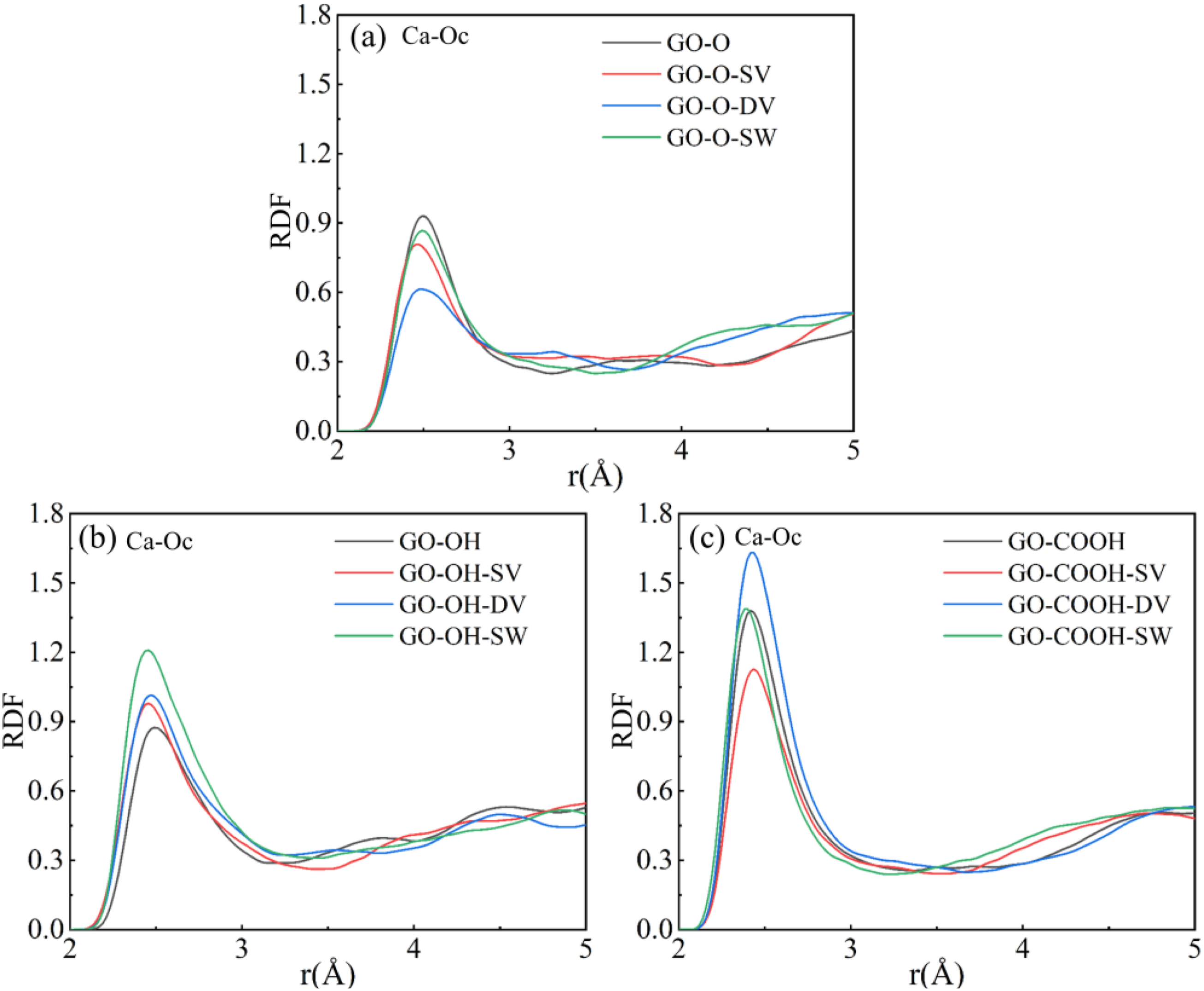
As shown by the radial distribution functions (RDFs) of Ca-Ow in the composites in Figure 10(a)–(d), the Gr (GO)/C-S-H composites all display a primary peak at approximately 2.44 Å, indicating the coordination of calcium ions between C-S-H layers with oxygen atoms in water. The insets reveal that defects cause a slight shift of the peak to the right and a small decrease in its intensity, with SW defects showing a more noticeable effect. Figure 11(a)–(c) demonstrates that in GO/C-S-H systems modified with different oxygen-containing functional groups, characteristic peaks for Ca-Ow coordination appear within the 2.44-2.52 Å range, directly illustrating the interfacial coordination of Ca2+ with oxygen atoms on the GO surface. Different types of defects affect the peak’s intensity and position, with higher peak values observed under DV defects, indicating that DV defects enhance the bonding between Ca2+ and oxygen atoms on GO sheets. Structurally and mechanistically, the DV defect removes two adjacent carbon atoms, restructuring the local bonding environment. This slightly alters the spatial arrangement and coordination geometry of nearby oxygen-containing groups. On one hand, the local distortion caused by the defect shortens some Ca-Oc bonds, increasing the compatibility of coordination between Ca2+ and oxygen atoms. On the other hand, changes in local bond angles and lengths result in a distribution of oxygen lone-pair electrons that favor stable Ca2+ coordination, as shown by a small increase in the RDF peak intensity. These defects minimally influence the Ca-Ow and Ca-Oc coordination environments. The minor shifts in RDF peak positions and slight variations in peak heights do not modify the core coordination structure and have limited impact on interfacial bonding and microstructure. (a)-(d) shows the RDF curves of the Gr (GO)/C-S-H composite Ca-Ow for complete, SV, DV, and SW defect types. (a)-(c) shows the RDF curves of the Ca-Oc phase in the GO/C-S-H composite for the complete, SV, DV, and SW defect types.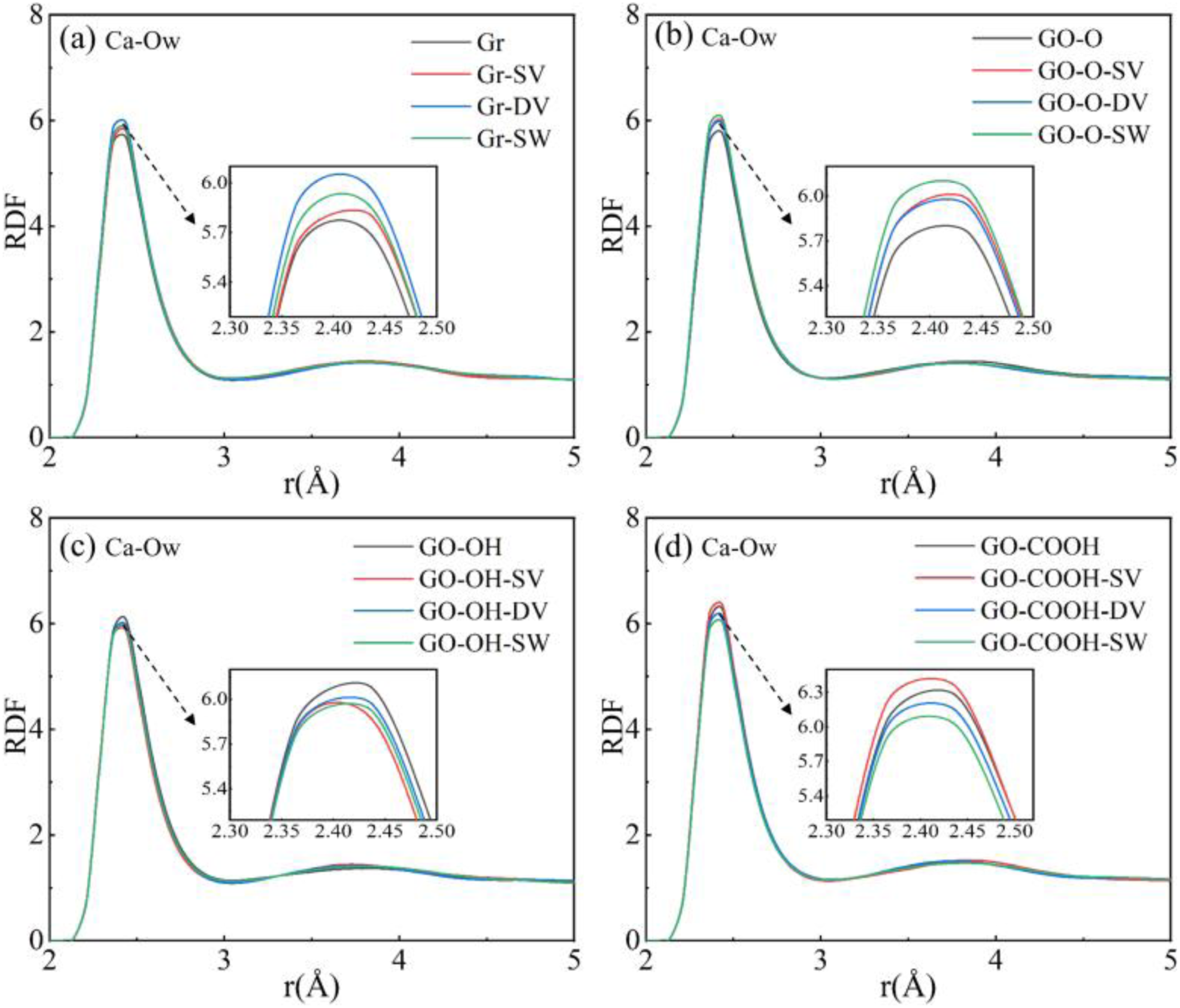
As can be seen from the mean square displacement (MSD) of C atoms in the composites shown in Figure 12(a)–(d), the MSD of C atoms in both the intact structure and the DV defect in Gr/C-S-H increases gradually. In contrast, the MSD significantly rises under SV and SW defects, with the fastest growth seen in SW defects. This suggests that topological defects and vacancies disturb the regularity of graphene’s sp2-hybridized carbon hexagonal rings, weakening interatomic bonds and boosting the mobility of carbon atoms. In GO-O/C-S-H, the overall MSD across different defects is lower than in Gr/C-S-H, but the increase under SW defects remains the most notable. This indicates that epoxy groups provide some interfacial constraint on atomic movement, though defects still enhance carbon atom diffusion. The MSD increase was most significant for SV defects in the GO-OH/C-S-H system and for DV defects in the GO-COOH system. This phenomenon demonstrates that hydroxyl and carboxyl groups can form strong hydrogen bonds with C-S-H, partially offsetting the weakening effect of defects on the carbon skeleton and thereby limiting excessive carbon atom migration. (a)-(d) shows the MSD curves of carbon atoms in the Gr (GO)/C-S-H composite for complete, SV, DV, and SW defect types.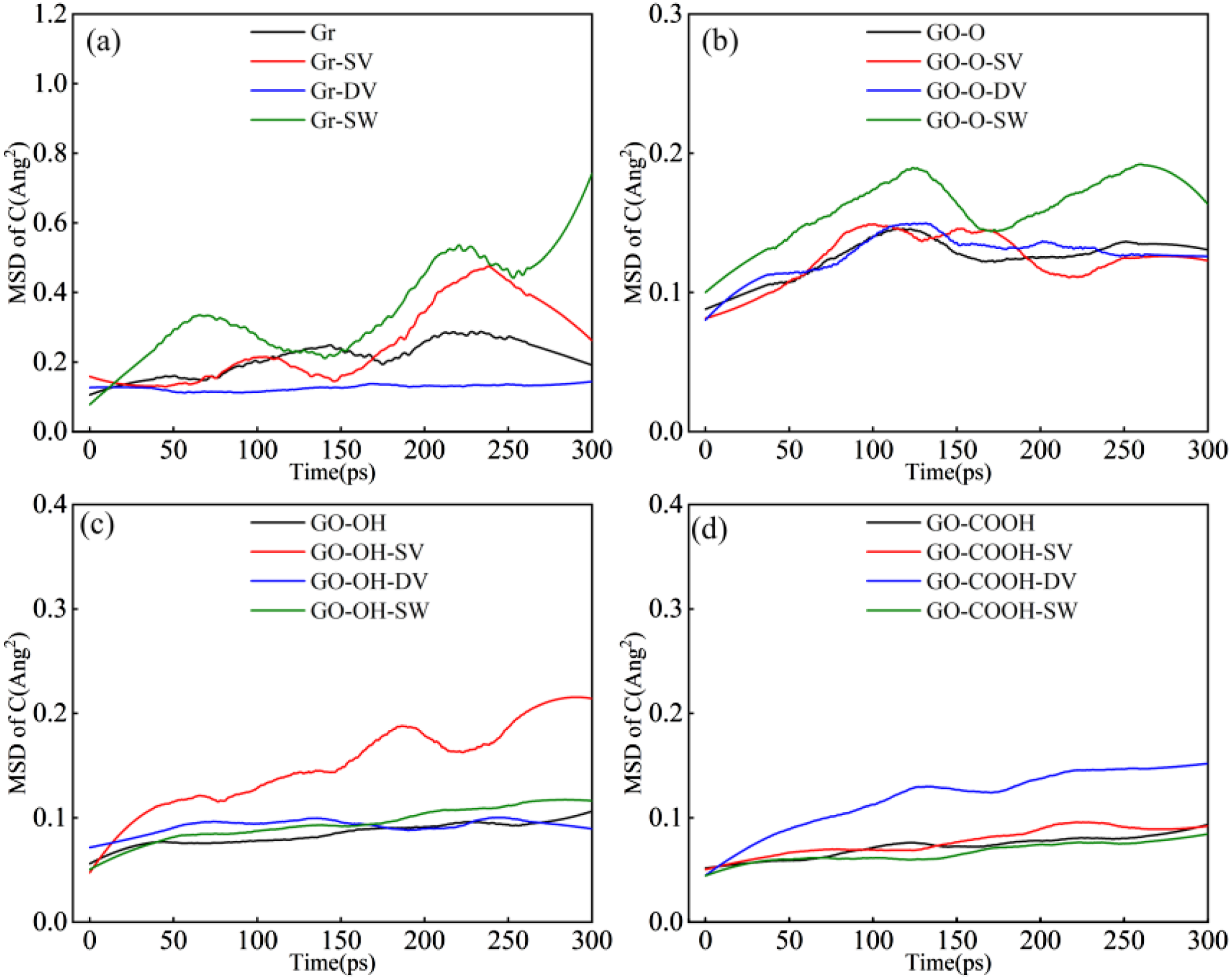
The tensile stress-strain curves shown in Figure 13(a)–(d) indicate that, following the introduction of defects, the peak stress and the slope of the elastic region for both Gr/C-S-H and GO-O/C-S-H decreased slightly. Defects directly disrupt the intrinsic sp2-hybridized planar hexagonal carbon ring structure, creating localized stress concentration points that weaken the inherent strength of the layers and reduce the efficiency of interfacial load transfer. Meanwhile, the Gr surface remains inert, bonding to the C-S-H matrix solely through van der Waals forces. The interfacial enhancement effect from increased surface-active sites caused by defects is insufficient to offset the destructive impact of defects on the carbon skeleton, resulting in an overall decline in performance. Interestingly, the peak stress of GO-OH/C-S-H and GO-COOH/C-S-H composites shows a slight increase after defect introduction, along with a marginal rise in the slope of the curve’s ascending segment. As illustrated in Figure 14(a)–(d), the tensile strengths of both the GO-OH-SV/C-S-H (2.59 GPa) and GO-COOH-SV/C-S-H (2.61 GPa) composites increase. This is due to the formation of strong hydrogen bonds between the hydroxyl and carboxyl groups and the C-S-H groups. The defects increase the number of surface-active sites and enhance interfacial bonding, thereby counteracting the weakening of the layered structure. The failure models in Figure 15(a)–(d) clearly indicate that at a strain of 0.4, failure in all Gr (GO)/C-S-H composites is mainly characterized by interfacial cracking along the z-direction, manifesting as delamination cracks between the flakes and the C-S-H matrix. The number of microcracks at defect sites in Gr/C-S-H is greater, with a wider propagation range, whereas cracks in GO/C-S-H are more focused and show no significant tendency to spread. This clearly demonstrates that the interfacial bonding strength between GO and the C-S-H matrix is superior. Even in the presence of defects, this bonding effectively suppresses microcrack propagation, thereby reinforcing the composite material. (a)-(d) shows the stress-strain curves of the Gr (GO)/C-S-H composite under z-direction tensile loading for complete, SV, DV, and SW defect types, respectively. (a)-(d) shows the tensile strength and Young’s modulus of the Gr (GO)/C-S-H composite along the z-direction for complete, SV, DV, and SW defect types. (a)-(d) Failure models of Gr (GO)/C-S-H composites under z-direction tensile loading for complete, SV, DV, and SW defect types at a strain of 0.4.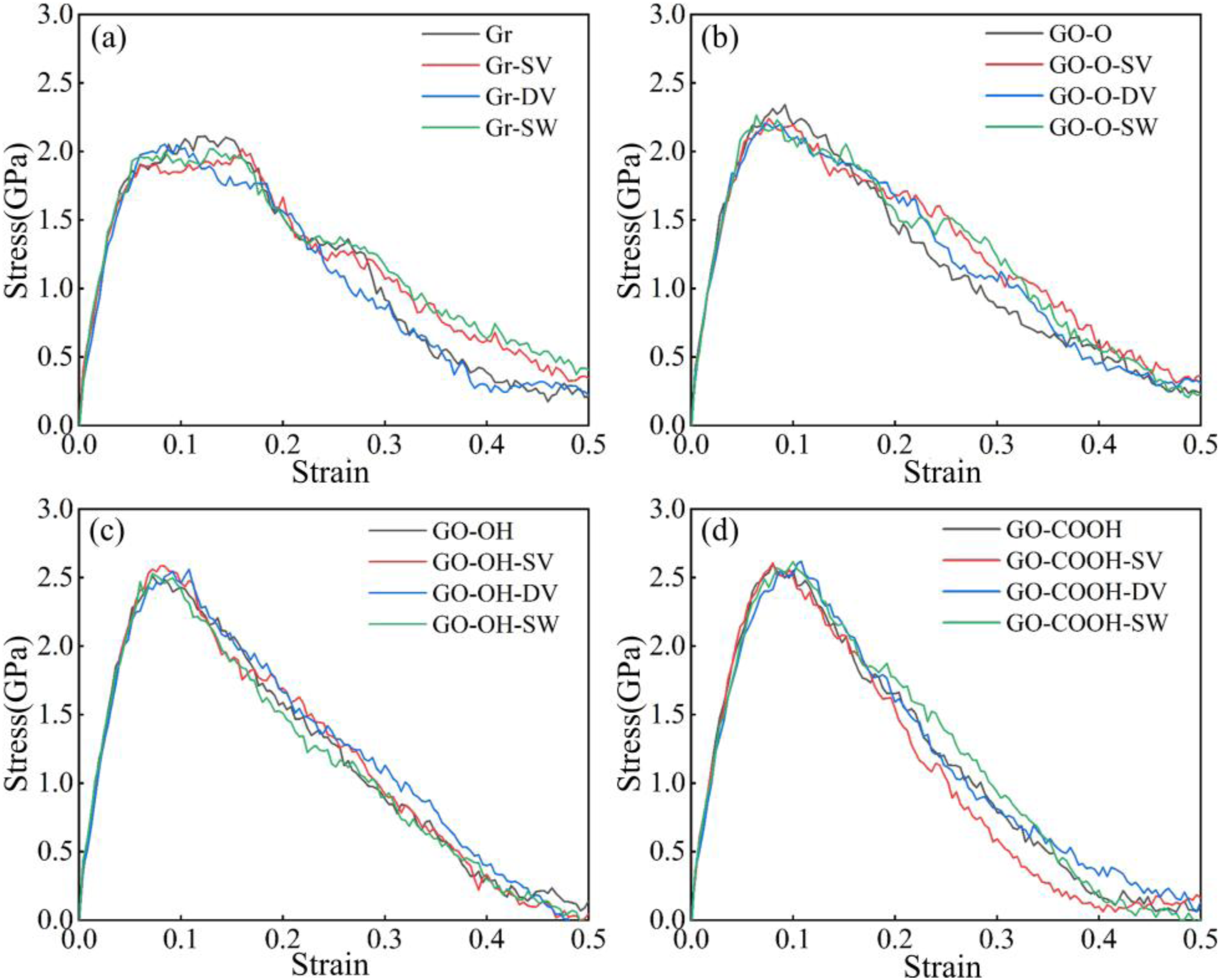
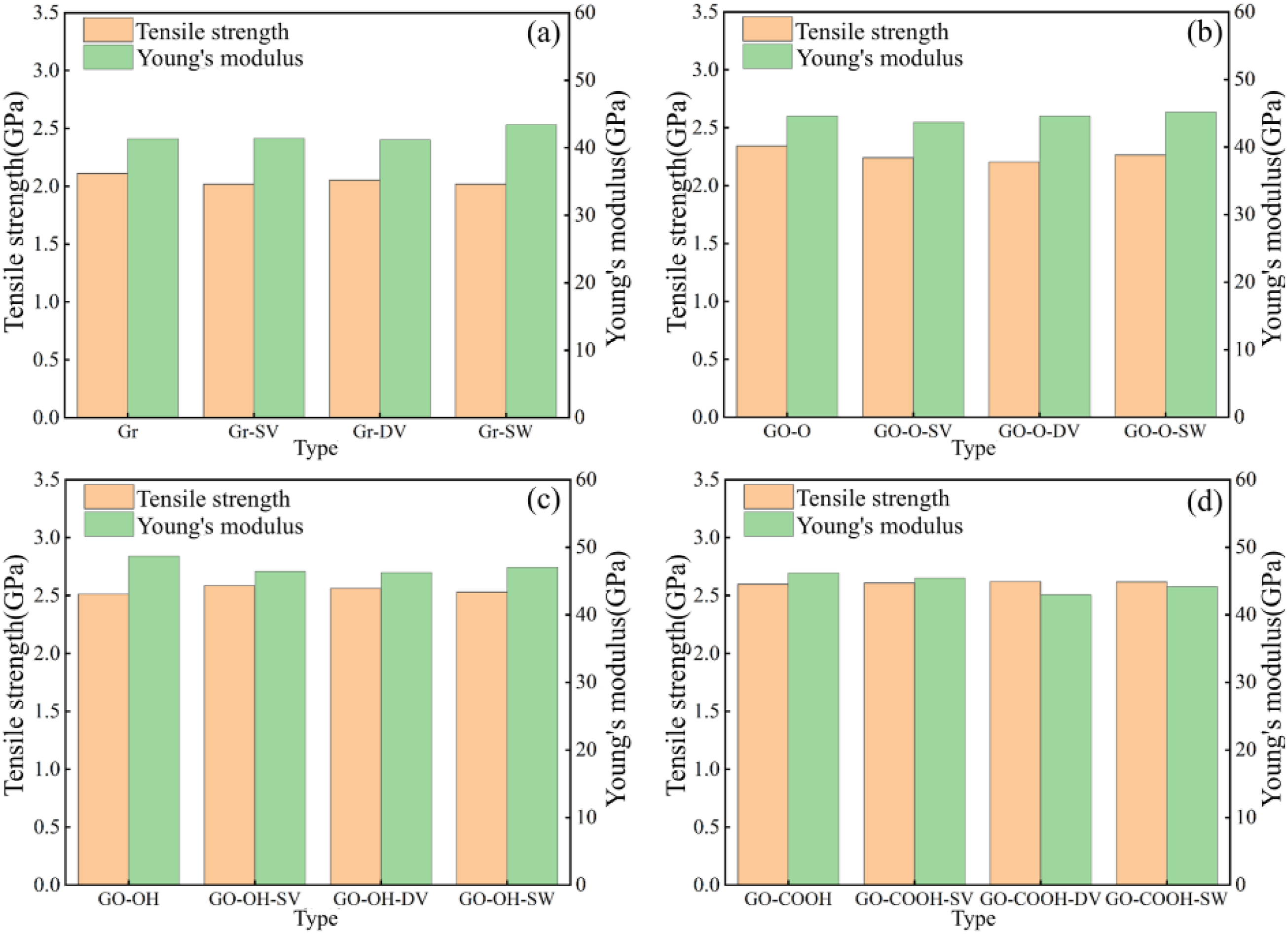
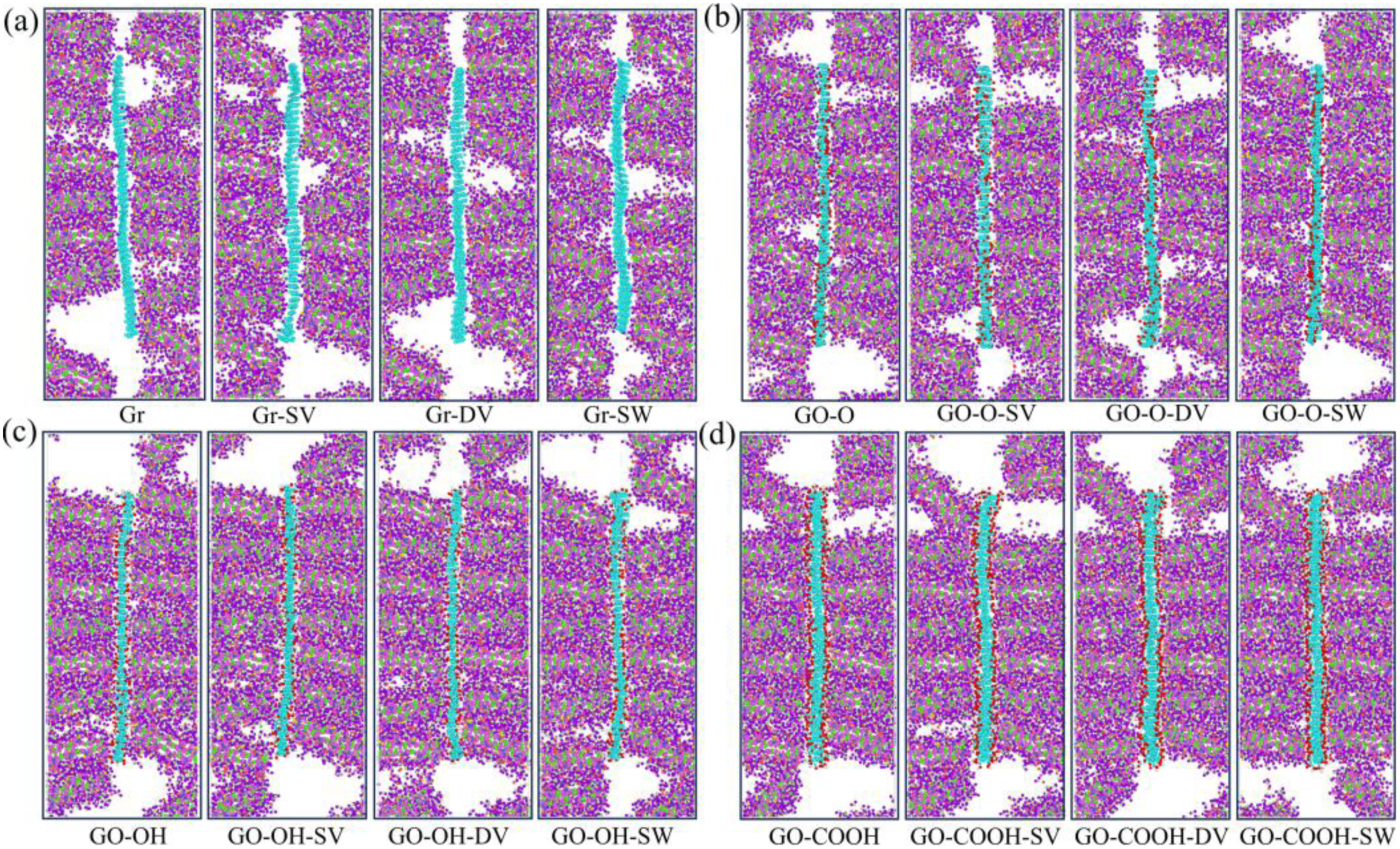
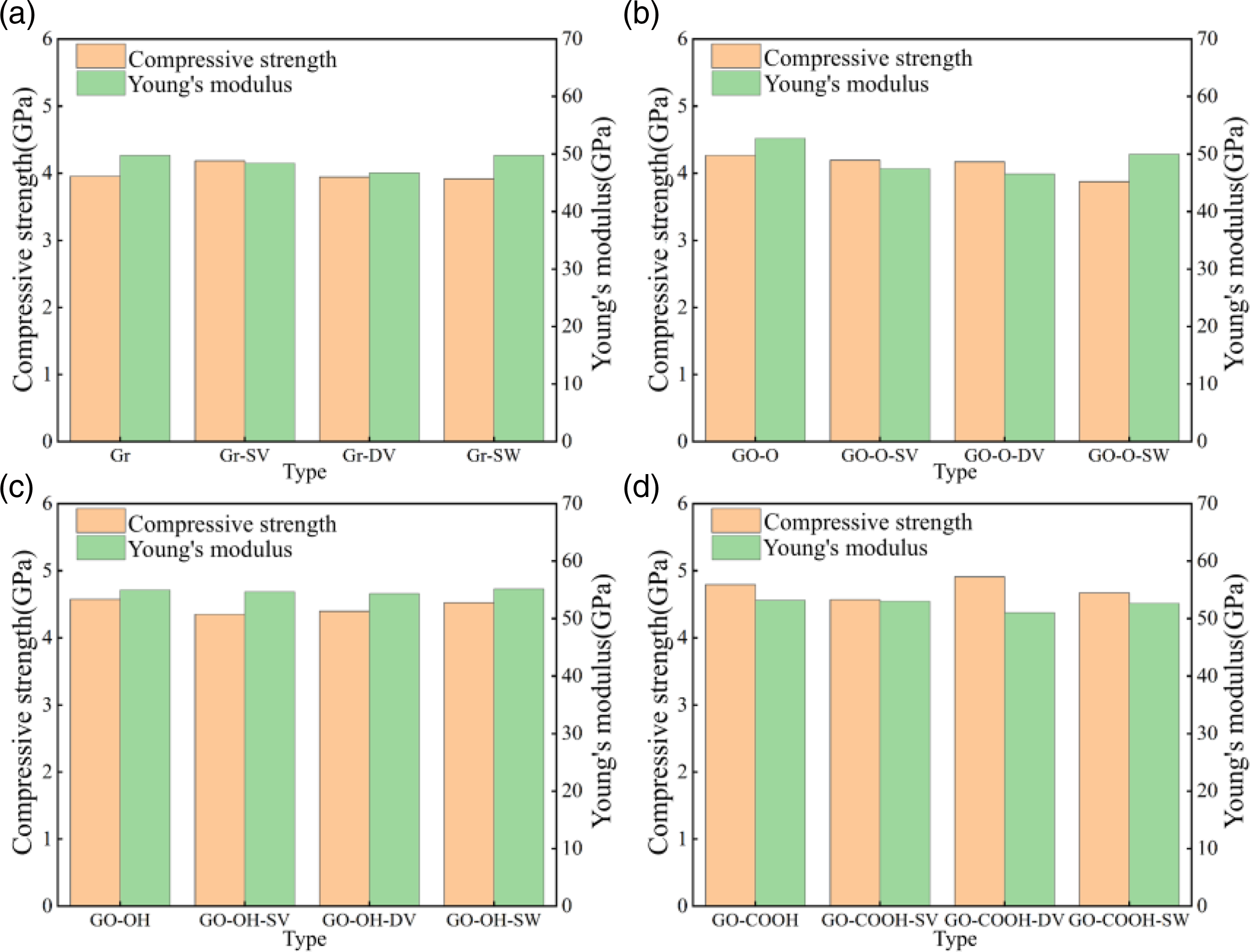
The compressive stress–strain curves shown in Figure 16(a)–(d) indicate that the stress in the elastic region of the Gr (GO)/C-S-H composite increases linearly with strain. The peak stress of Gr (GO)/C-S-H composites is relatively unaffected by defects, exhibiting only slight fluctuations. The presence of oxygen-containing functional groups enhances the disruptive effect of defects on the material’s mechanical properties. As shown in Figure 17(a)–(d), the compressive strength of Gr/C-S-H slightly rises with SV defects (4.19 GPa) but declines with DV and SW defects. This could be because defects in the silicon wafer create localized stress relief points by marginally altering the layered structure. The compressive strength of GO-O/C-S-H generally diminishes across the three defect types, indicating that the material’s susceptibility to defects considerably increases after epoxy group modification. The compressive strength of GO-OH/C-S-H and GO-COOH/C-S-H shows irregular changes depending on the defect type. The behavior of Young’s modulus is even more complex; in some cases, the Young’s modulus of defective Gr (GO)/C-S-H composites is slightly higher than that of undamaged ones. Under compressive load, the localized stress concentrations caused by defects can be reduced through sheet slip and matrix deformation. (a)-(d) shows the stress-strain curves of the Gr (GO)/C-S-H composite under z-direction compression for complete, SV, DV, and SW defect types, respectively. (a)-(d) shows the compressive strength and Young’s modulus of the Gr (GO)/C-S-H composite along the z-direction under complete, SV, DV, and SW defect types.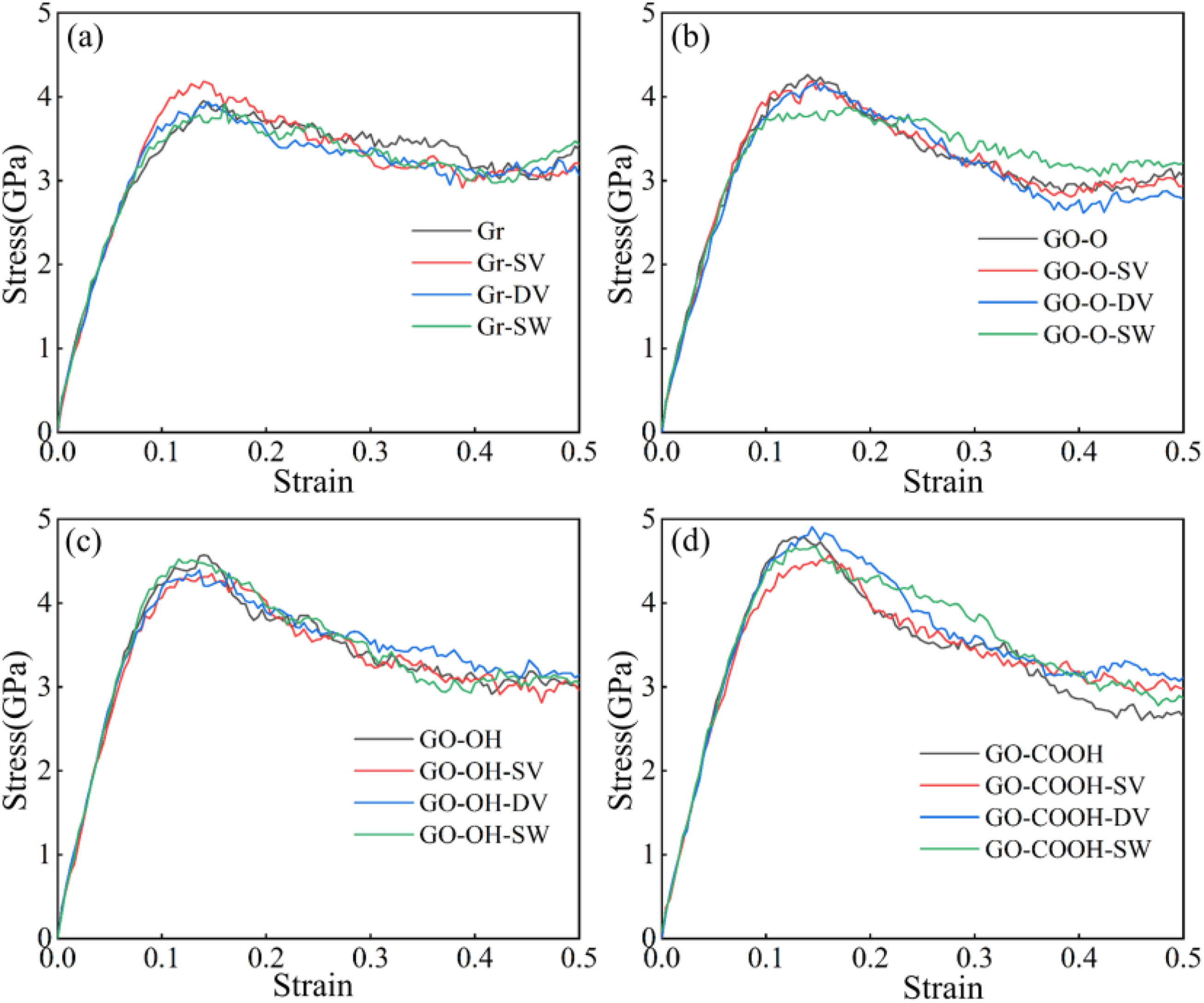
Effect of temperature on the interface structure and mechanical properties of Gr/GO and C-S-H composites
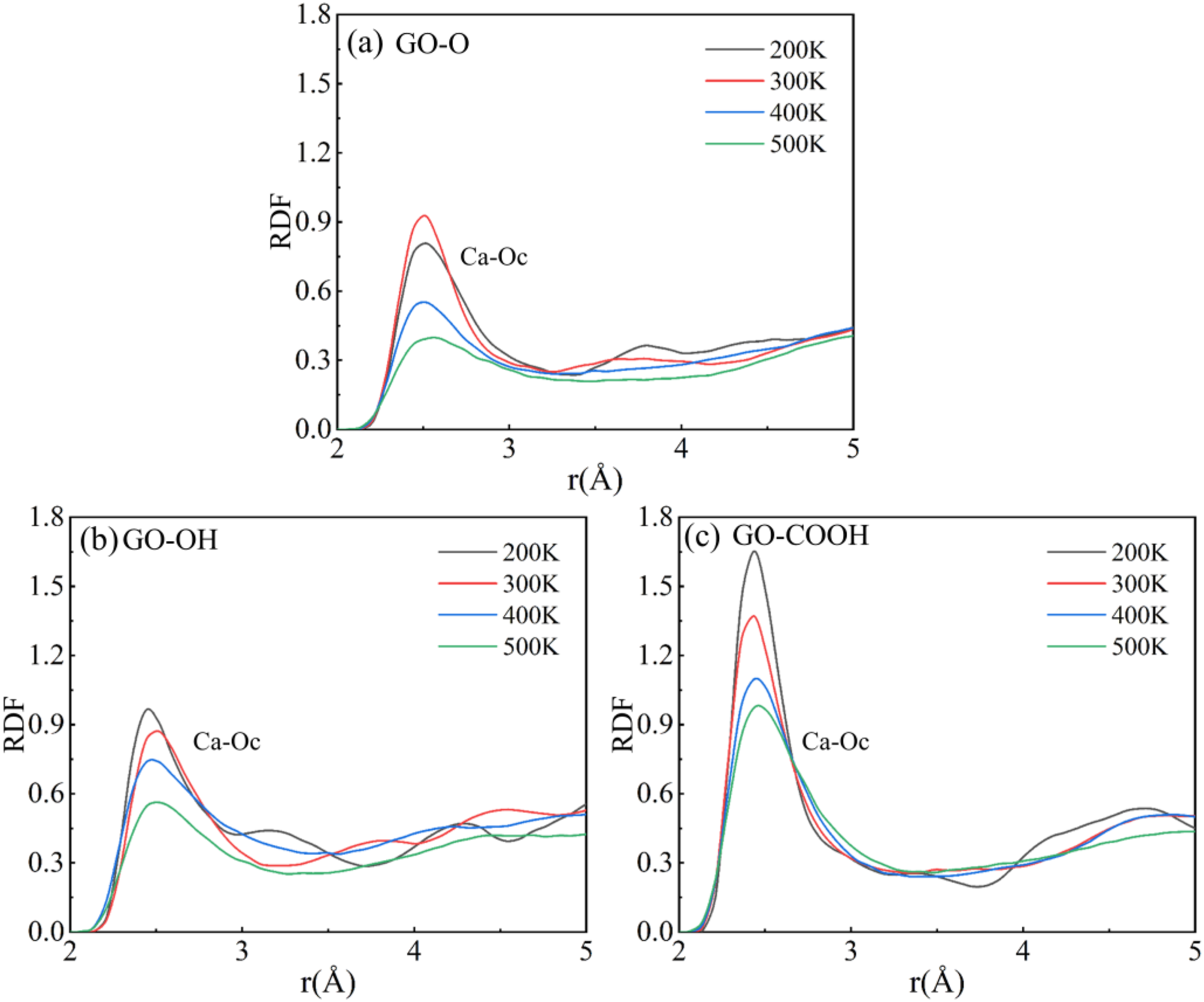
The RDF results shown in Figure 18(a)–(d) and Figure 19(a)–(c) clearly illustrate how temperature impacts the interfacial coordination structure. In each system, the Ca-Ow characteristic peak occurs at about 2.44 Å, while the Ca-Oc characteristic peak in GO/C-S-H is spread across 2.44-2.52 Å. This corresponds to the first coordination sphere of Ca2+ with water molecules in the C-S-H interlayer and with surface oxygen atoms on the GO surface, respectively. As temperature increases from 200 K to 500 K, both types of coordination peaks sharply decrease in intensity, broaden significantly, and shift in position. The main reason is that the atoms’ thermal vibrational energy rises sharply at high temperatures, which not only disrupts electrostatic attraction and the stability of Ca2+ coordination bonds with oxygen atoms, but also disturbs the ordered arrangement of atoms at the interface. This results in a reduction of local coordination density and a more spread-out distribution of bond lengths. Simultaneously, thermal motion weakens the interaction between Ca2+ and Ow (Oc), increasing bond distance and decreasing bond energy. This directly weakens interfacial bonding and reduces the orderliness of the microstructure, ultimately acting as a key microscopic factor in the ongoing decline of the composite material’s high-temperature mechanical properties. (a)-(d) shows the RDF curves of the Gr (GO)/C-S-H composite Ca-Ow at different temperatures. (a)-(c) shows the RDF curves of Ca-Oc in GO/C-S-H composites at different temperatures.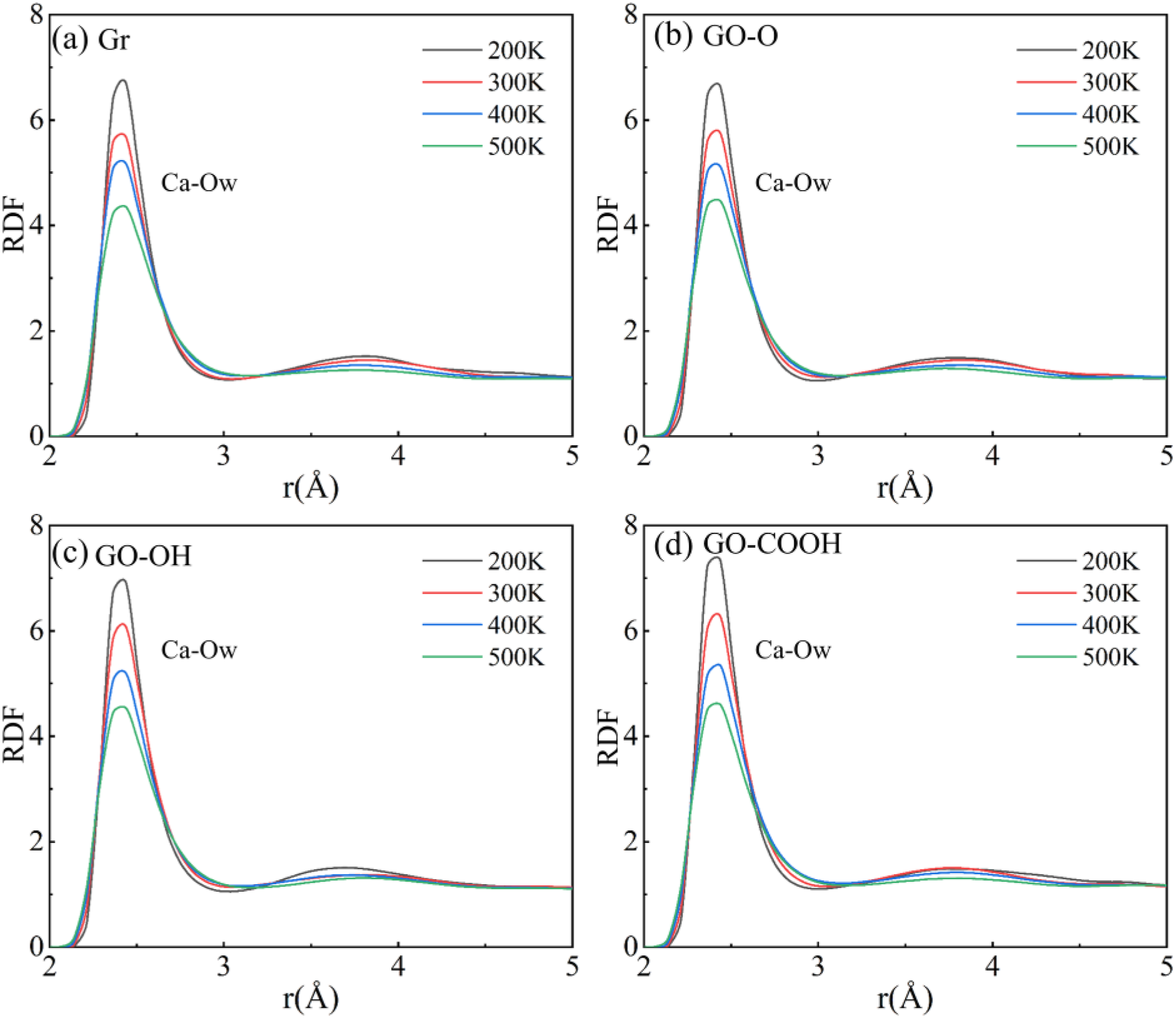
As shown by the MSD in Figure 20(a)–(d), increasing temperature provides atoms with more thermal kinetic energy, causing localized vibrations of carbon atoms to gradually shift from equilibrium positions to long-range diffusion, as indicated by the continuous rise in MSD with increasing temperature. At 500 K, thermal motion’s energy is enough to significantly weaken interfacial constraints, leading to a sharp increase in carbon atom mobility. Among these, Gr/C-S-H and GO-O/C-S-H show the weakest interfacial constraints due to only weak van der Waals forces or weak epoxide coordination, resulting in the most dramatic rise in MSD. Conversely, the GO-OH/C-S-H and GO-COOH/C-S-H interfaces, stabilized by strong coordination bonds between hydroxyl and carboxyl groups and C-S-H groups, along with a hydrogen bond network, effectively restrict interfacial carbon atoms and greatly slow their thermal diffusion. Consequently, the MSD increases much more slowly at higher temperatures. These findings suggest that the robust interfacial bonds formed by oxygen-containing functional groups are vital for improving the structural stability of composite materials at elevated temperatures. (a)-(d) shows the mean square displacement (MSD) curves of carbon atoms in Gr (GO)/C-S-H composites at different temperatures.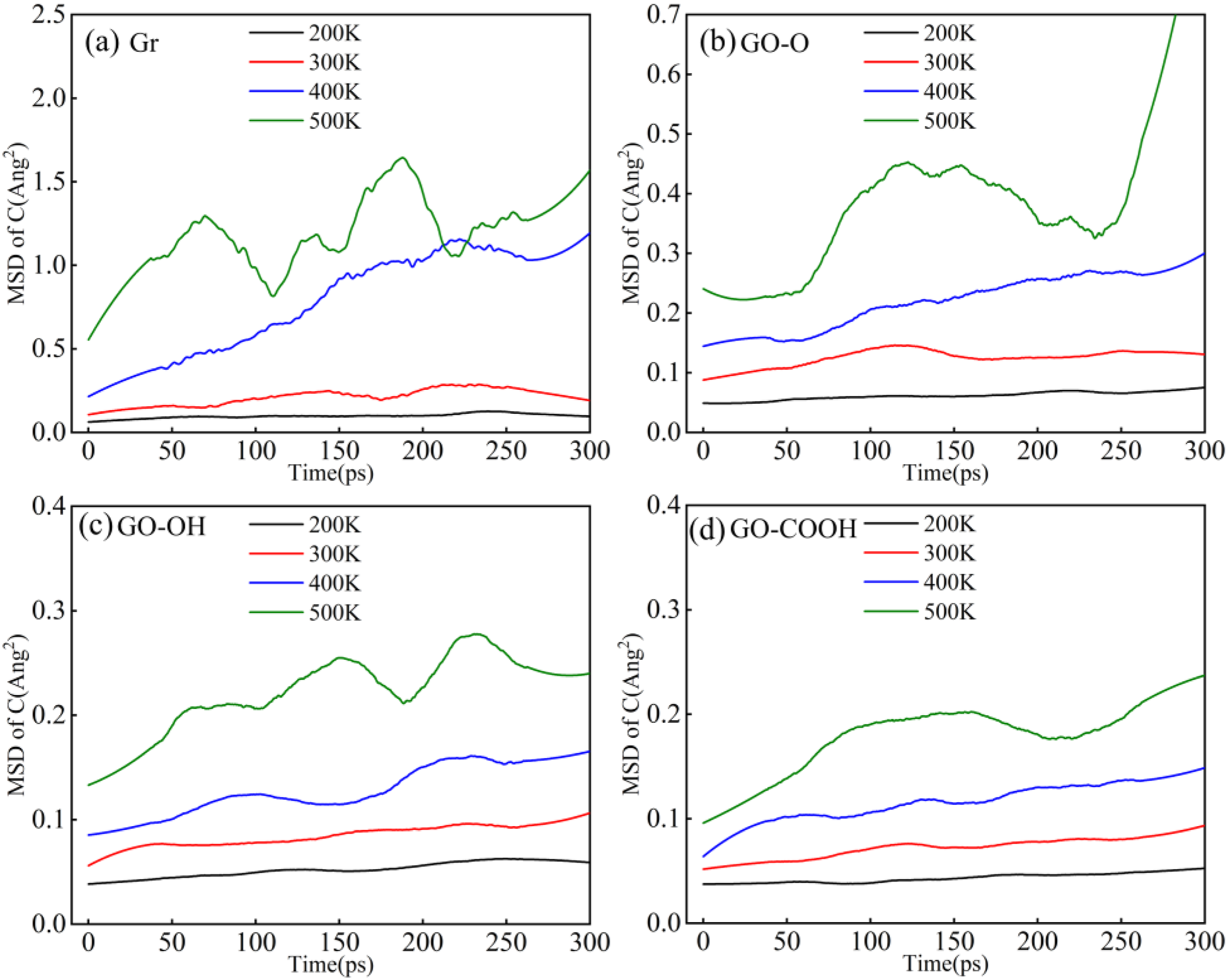
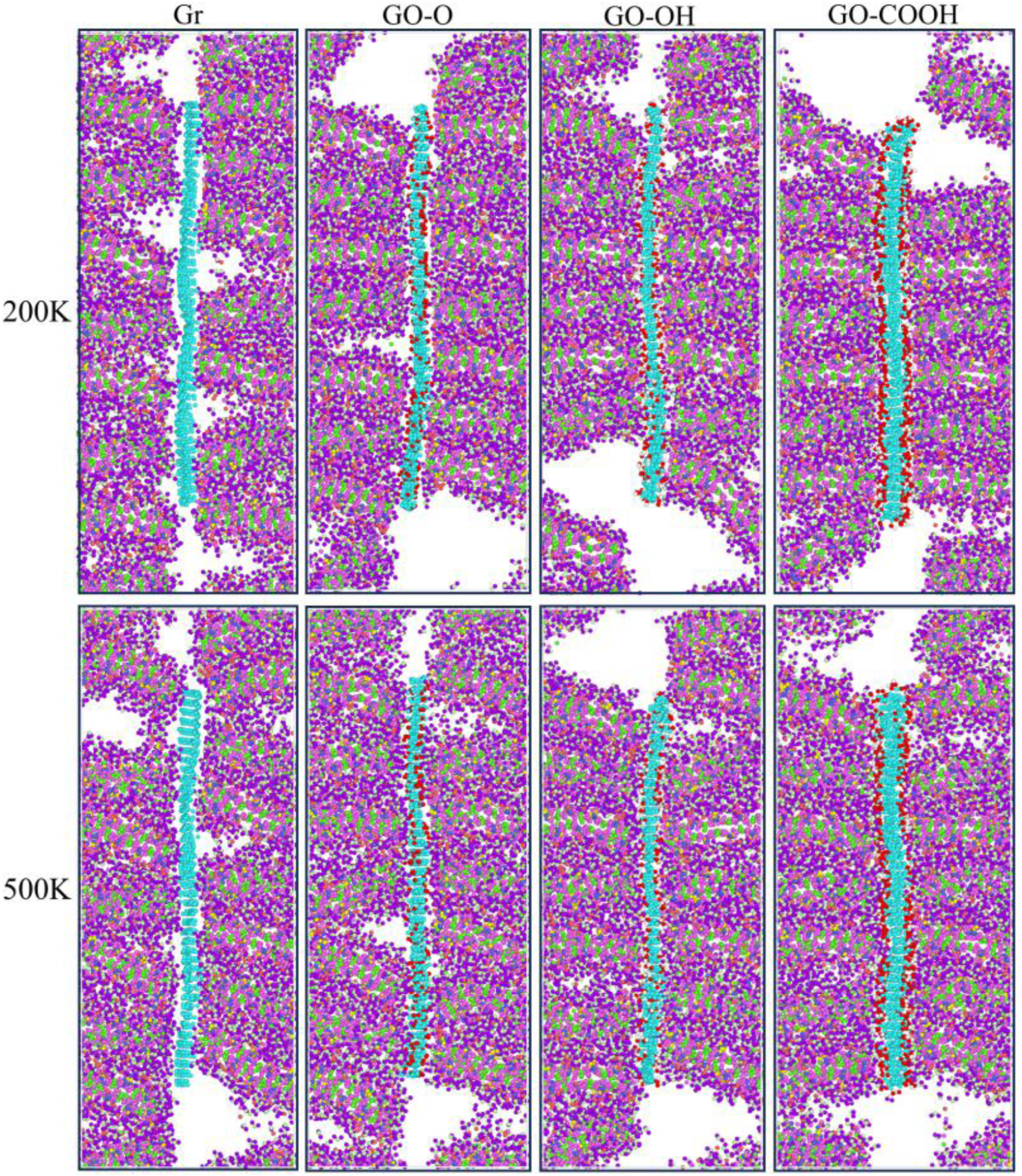
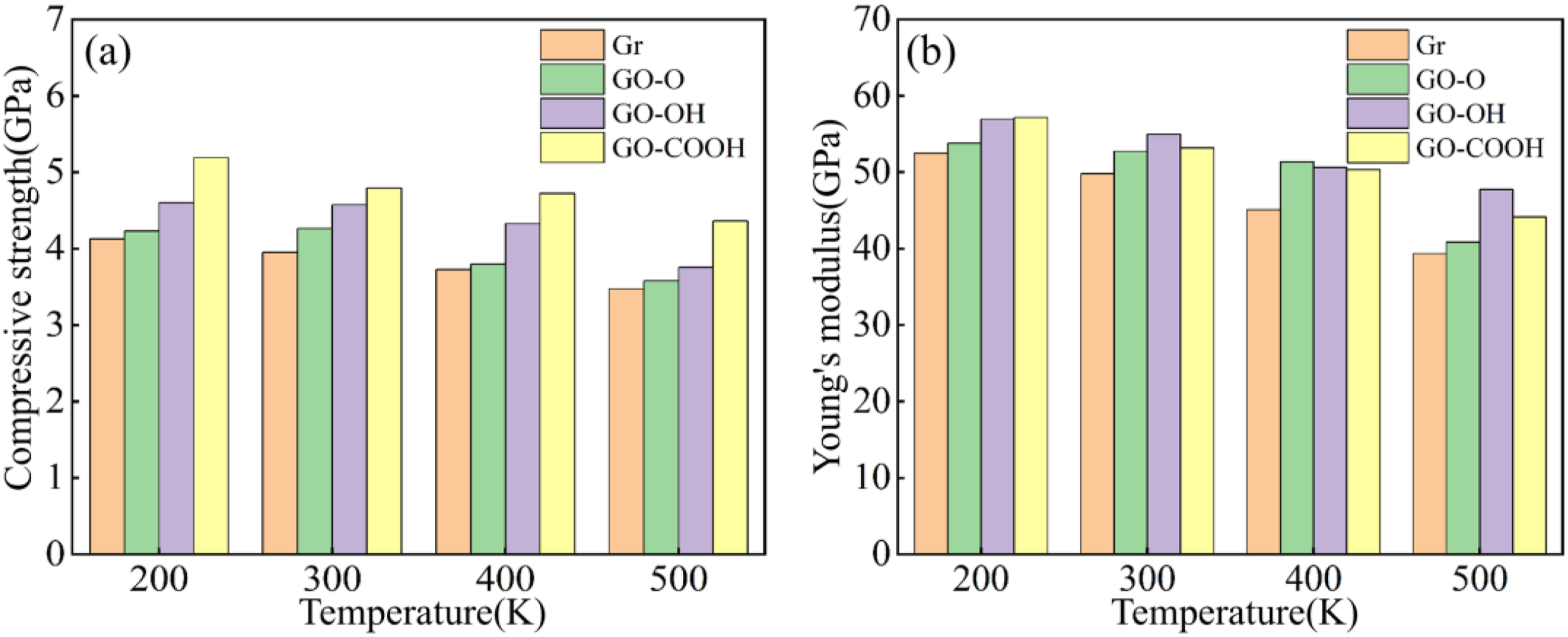
The tensile mechanical results shown in Figure 21(a)–(d) and Figure 22(a)–(b) indicate that stress increases linearly during the low-strain region, peaks at about 0.1 strain, and then enters the plastic region. As temperature rises, tensile strength and Young’s modulus decrease monotonically. This occurs because high temperatures amplify atomic thermal motion, disrupting interfacial coordination bonds and hydrogen bond networks, which weakens the efficiency and strength of interfacial load transfer. At 200 K, both tensile strength and Young’s modulus of GO-OH/C-S-H reach their highest values. At 500 K, GO-COOH/C-S-H still maintains the highest tensile strength, and the reduction in modulus for both GO-OH/C-S-H and GO-COOH/C-S-H is notably smaller. This confirms that a stable interface formed by hydroxyl and carboxyl groups through strong coordination and hydrogen bonding is the key mechanism behind the enhanced tensile properties and high-temperature structural stability of the composite materials. The microstructural failure morphology shown in Figure 23 reveals the mechanisms behind performance changes. At 200 K, the interface bonding is strong, with failure mainly caused by localized delamination and microcracks. At 500 K, interfacial bonding weakens due to increased thermal motion, leading to more evident delamination and matrix cracking. Notably, interfacial delamination of Gr/C-S-H and GO-O/C-S-H is most prominent, while the interfaces of GO-OH/C-S-H and GO-COOH/C-S-H remain relatively intact. (a)-(d) shows the stress-strain curves of the Gr (GO)/C-S-H composite material under z-direction tensile loading at different temperatures. (a) and (b) show the tensile strength and Young’s modulus of Gr (GO)/C-S-H composites along the z-direction at different temperatures, respectively. Failure models of Gr (GO)/C-S-H composites under z-direction tensile loading at temperatures of 200 K and 500 K when the strain reaches 0.4.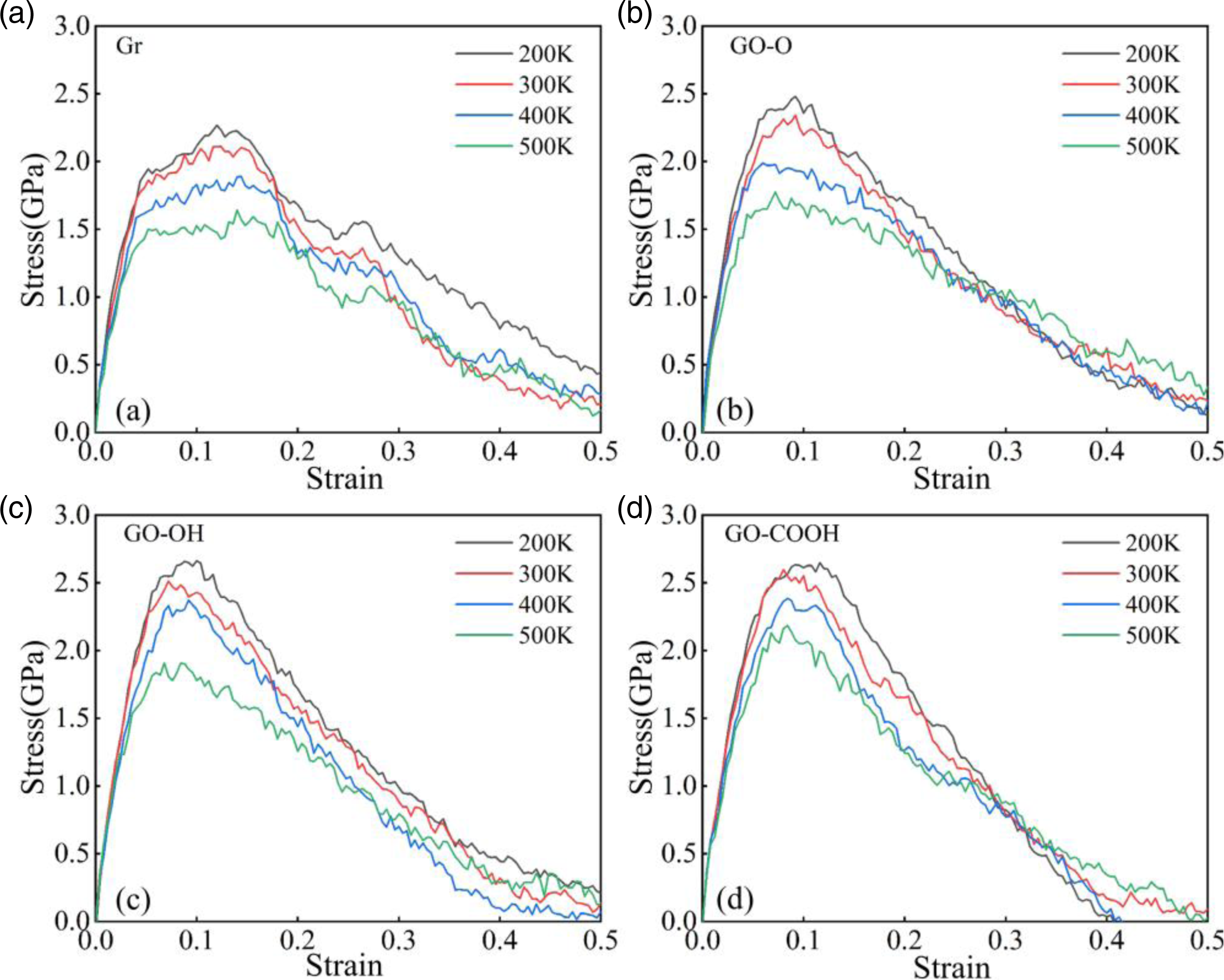
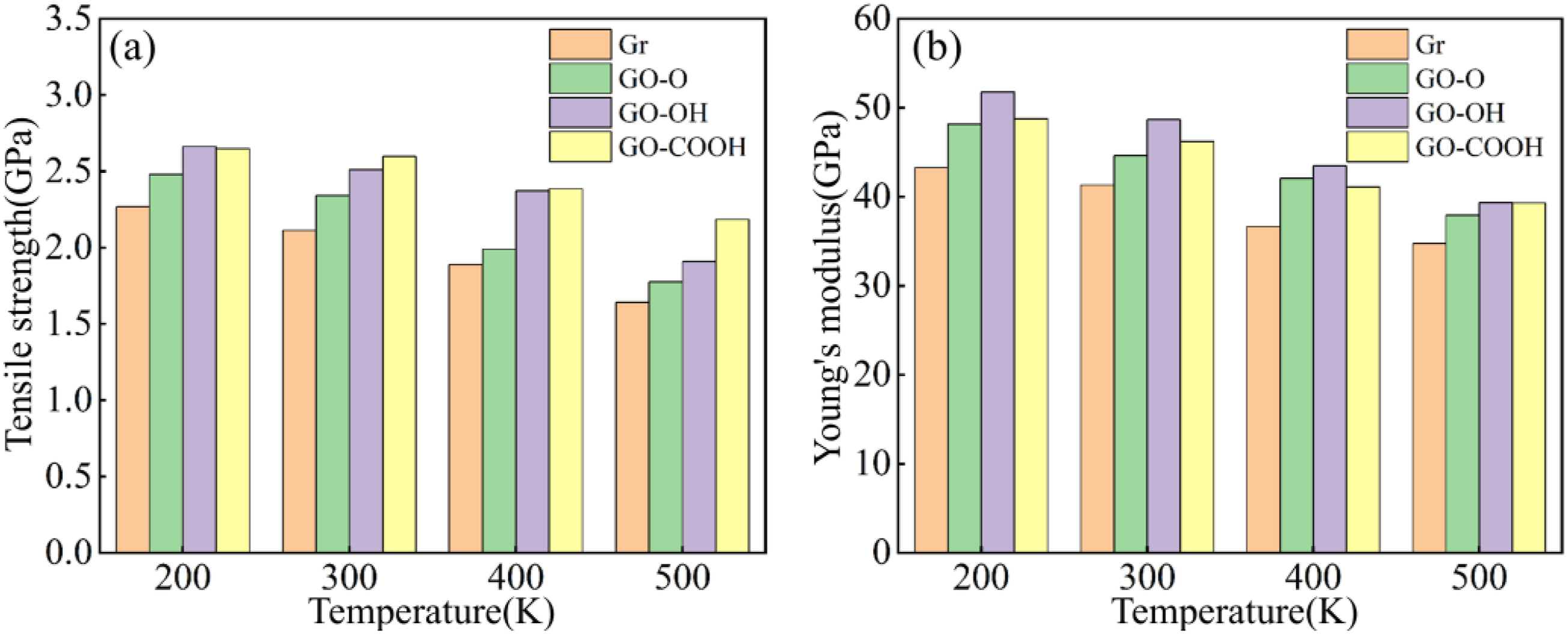
The compression mechanics results shown in Figure 24(a)–(d) and Figure 25(a)–(b) indicate that, as temperature increases, both the peak stress and the slope of the elastic region in the composite material decrease significantly, while the compressive strength and Young’s modulus exhibit a monotonically decreasing trend. This phenomenon is fundamentally linked to the weakening of internal bonding within the material at high temperatures; high temperatures disrupt the interfacial bonding between Gr (GO) and the C-S-H matrix, while simultaneously increasing the number of lattice defects in the C-S-H matrix itself, thereby reducing the composite material’s compressive strength and stiffness. At a low temperature of 200 K, the GO-COOH/C-S-H composite exhibits the highest compressive strength and Young’s modulus. This is primarily attributed to the stronger hydrogen bonding formed between the carboxyl functional groups on the GO-COOH surface and the C-S-H matrix, which effectively enhances the interfacial bonding strength. The GO-OH/C-S-H composite exhibits the next-highest performance; the hydroxyl functional groups still promote interfacial interactions, significantly outperforming the Gr and GO-O systems. When the temperature rises to 500 K, GO-COOH/C-S-H still exhibits the highest compressive strength, owing to the superior thermal stability of the carboxyl-Ca2+ coordination bonds. In contrast, the modulus of GO-OH/C-S-H surpasses that of GO-COOH/C-S-H, which can be attributed to the hydroxyl groups’ greater tendency to form a continuous cross-linked network with the C-S-H siloxane backbone at high temperatures, thereby maintaining higher deformation stiffness through interfacial structural rearrangement. (a)-(d) shows the stress-strain curves of Gr (GO)/C-S-H composites compressed along the z-direction at different temperatures. (a) and (b) show the compressive strength and Young’s modulus of Gr (GO)/C-S-H composites compressed along the z-direction at different temperatures, respectively.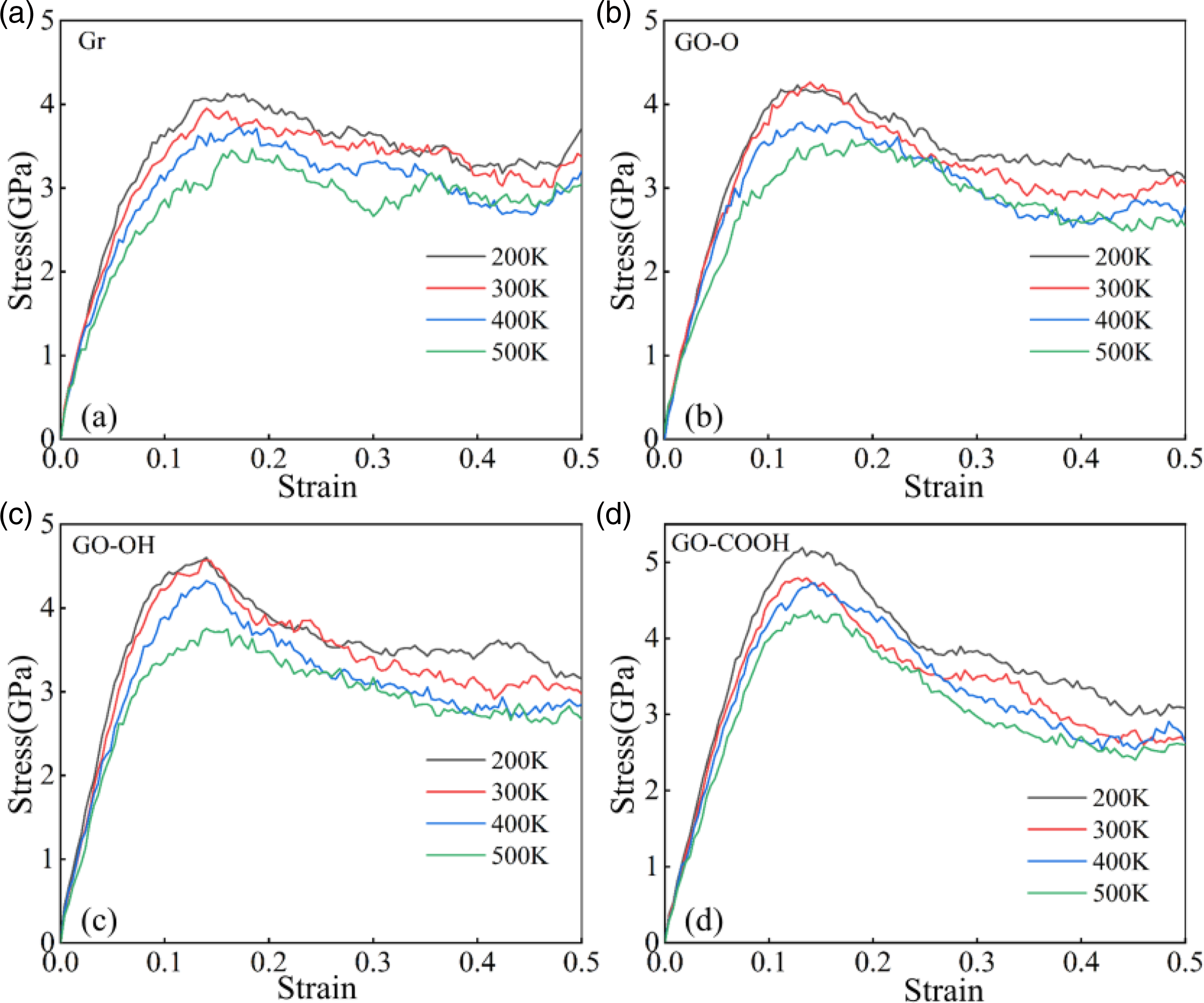
Conclusion
This study employs molecular dynamics simulations to investigate the effects of functional groups, defects, and temperature on the interfacial structure and mechanical properties of Gr (GO)/C-S-H composites. The core conclusions are as follows. (1) Experimental results show that both Gr and GO can improve the compressive and flexural strengths of cement-based composites, with GO providing better reinforcement effects. The reference compressive strength of standard cement mortar is 42.57 MPa, and its flexural strength is 6.76 MPa. Gr performed best at a dosage of 0.075%, increasing compressive and flexural strengths by 6.6% and 31.4%, respectively. GO showed the best results at a dosage of 0.03%, boosting compressive and flexural strengths by 14.3% and 34.3%, respectively. At this optimal dosage, its strength surpassed that of Gr-modified specimens. Simulation results suggest that GO’s oxygen-containing functional groups enhance the material’s properties by forming stable coordination bonds with Ca2+ through carboxyl and hydroxyl groups. GO-COOH/C-S-H achieves the highest tensile and compressive strengths, while GO-OH/C-S-H exhibits the highest Young’s modulus. (2) Temperature is the main factor affecting the properties of composite materials. As the temperature rises from 200K to 500K, the connection between Ca and Ow(Oc) weakens considerably, and the rate of C atom migration greatly increases. This results in a steady decline in tensile strength, compressive strength, and Young’s modulus across all systems, with Gr/C-S-H and GO-O/C-S-H systems showing more pronounced performance loss. Modifications with hydroxyl and carboxyl groups effectively reduce high-temperature degradation, helping the composite material retain better thermal stability. (3) At a defect concentration of 2%, the effects of single vacancies (SV), double vacancies (DV), and topological defects (SW) on the interfacial structure and mechanical properties of composite materials are considerably less significant than those of functional groups and temperature. Regarding interfacial structure, defects only cause minor fluctuations in the Ca-Ow and Ca-Oc characteristic peaks without substantially altering the fundamental coordination structure. Tensile strength slightly decreased for the Gr/C-S-H and GO-O/C-S-H composites, while it slightly increased for the GO-OH/C-S-H and GO-COOH/C-S-H composites. Under compression, compressive strength and Young’s modulus fluctuated within a narrow range, with some composites showing slight performance improvements.
This study has clarified the primary regulatory influence of temperature on composite material properties, offering a crucial theoretical foundation for enhancing the mechanical properties and high-temperature stability of cementitious composites through functional group modification.
Footnotes
Authors Contributions
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research is financially supported by the National Natural Science Foundation of China (Grant No. 52350410476 and 52361135807), the Department of Science and Technology of Shandong Province (Grant No. 2021CXGC011204). and the Liaoning provincial key laboratory of Safety and Protection for Infrastructure Engineering.
Data Availability Statement
Data will be made available on request.