
Abstract
The ternary superparamagnetic nanocomposites consisting of graphene oxide (GO), Fe3O4 nanoparticles, and optically active poly(amide-imide) (PAI) were fabricated in three steps consisting of a facile one-pot in situ growth of Fe3O4 on GO, resulted in the preparation of the magnetic Fe3O4@GO, modification of Fe3O4@GO by 3-aminopropyltriethoxy silane to introduce amino groups on its surface, and subsequently its compositing by various levels of 5, 10, and 15 wt% with chiral PAI derived from 3,5-diamino-N-(4-(di(1H-indol-3-yl)methyl)phenyl)benzamide and N,N′-(4,4′-carbonyldiphthaloyl)-bis-
Introduction
Graphene oxide (GO) as a promising nanomaterial has attracted substantial interest owing to its exceptional structural, mechanical, electronic, and thermal properties and extensive range of potential applications, such as catalysts, drug delivery, supercapacitors, biosensors, fuel cells, and so on.1–9 The magnetic nanoparticles are attractive in an extensive range of biomedical applications because these nanoparticles can be easily separated using an external magnetic field and redispersed quickly after removing the magnetic field.10–15 Moreover, the incorporation of magnetic nanoparticles into GO brings about GO-based magnetic nanocomposites with fascinating properties of two component materials for a variety of applications.16–21
Among the high-performance materials, aromatic polyimides (PIs) have been attracting extraordinary attention owing to their excellent performance, mechanical and electrical properties, and chemical and thermal stability. 22 However, these polymers are intractable as a result of their infusibility and insolubility in organic solvents. To overcome this problem, different co-PIs have been synthesized. Poly(amide-imide)s (PAI)s are one of the significant classes of high-performance co-PIs due to a positive synergy of characteristics from both PIs and polyamides, such as high strength, good solubility in organic solvents, excellent thermal stability, and high chemical resistance.23–28 These copolymers are the ideal candidate as matrix polymers for the advanced applications, such as sensors, electrical wire enamel, adhesives, membranes for separation, extrusion products, injection molding, and so on.29–32
In this study, ternary superparamagnetic nanocomposites of PAI/Fe3O4@GO were prepared using a simple and facile process. The synthesized nanocomposites were characterized by Fourier transform infrared (FTIR), X-ray diffraction (XRD), vibrating sample magnetometer (VSM), scanning electron microscope (SEM), and thermogravimetric analysis (TGA). Then, the sorption properties of Hg2+ by PAI/Fe3O4@GO 10 wt% are reported.
Experiment
Materials
All chemicals were purchased from Fluka Chemical Co. (Buchs, Switzerland), Aldrich Chemical Co. (Milwaukee, Wisconsin, USA), Riedel-de Hagen AG (Seelze, Germany), and Merck Chemical Co. (Germany). 3,3′,4,4′-Benzophenonetetracarboxylic dianhydride (Merck Chemical Co.) was purified with acetic anhydride in boiling acetic acid. L-Phenylalanine, acetic acid (glacial, 100%), indole, 4-nitrobenzaldehyde, ammonium acetate, hydrazine monohydrate, 10% palladium on activated carbon (Pd/C), 3,5-dinitrobenzoyl chloride, tetrabutylammonium bromide (TBAB), ethanol, iron (III) chloride hexahydrate, iron (II) chloride tetrahydrate, ammonium hydroxide, sulfuric acid (95–97%), graphite powder, potassium permanganate, hydrogen peroxide (30%), hydrochloric acid (37%), triphenyl phosphite (TPP), and 3-aminopropyltriethoxysilane (APTES, KH550) were purchased from Merck Chemical Co. and used as obtained without further purification.
Techniques
Inherent viscosities were measured by using a Cannon-Fenske Routine Viscometer (Germany) at a concentration of 0.5 g dL−1 at 25°C. Specific rotations were measured by a Jasco Polarimeter (Japan). FTIR spectra were recorded with a Jasco-680 Spectrometer (Japan) in the range of 400–4000 cm−1. Vibration bands were reported as wavenumber (cm−1). FTIR spectra of all samples were collected by making their pellets in potassium bromide (KBr) as a medium. The diffraction pattern of related materials was recorded in the reflection mode using a D8 ADVANCE Diffractometer, Bruker (Karlsruhe, Germany). Nickel-filtered copper Kα radiation (radiation wavelength, λ = 0.154 nm) was produced at an operating voltage of 45 kV and a current of 100 mA. The reactions were carried out on a Misonix Ultrasonic Liquid Processor, XL-2000 Series (Raleigh, North Carolina, USA). Ultrasound was a wave of frequency 2.25 × 104 Hz and a power of 100 W. TGA is performed with a STA503 Win TA at a heating rate of 10°C min−1 from 25°C to 800°C under nitrogen atmosphere. The magnetic properties were analyzed with a VSM (LDJ 9600 -1, Troy, Michigan, USA).
Adsorption investigations
For solid–liquid extraction of Hg2+, 5 mg of the nanocomposite was shaken with 25 mL of an aqueous solution of the metal salt with initial concentration of 10 mg L−1 for 3 days at 25°C (pH = 7). After centrifugal separation, the metal ion concentration in the supernatant was determined by atomic absorption spectroscopy. The removal capacity (qe, mg g−1) and the removal percentage (R%) of each metal ion were calculated using the following equations:

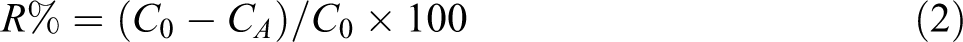
where V (L) is the volume of the solution, C0 and CA are the concentrations of metal ion in the initial solution and in the aqueous phase after adsorption, respectively, and W (g) is the amount of nanocomposite.
Synthesis of N,N′-(4,4′-carbonyldiphthaloyl)-bis-l -phenylalanine diacid (3)
To a 25-mL round-bottomed flask, 0.10 g (0.31 mmol) of 3,3′,4,4′-benzophenonetetracarboxylic dianhydride (1), 0.10 g (0.61 mmol) of L-phenylalanine (2), and 2 mL of acetic acid were placed. The mixture was stirred at room temperature for 8 h and then refluxed for 10 h. The reaction mixture was poured into a mixture of concentrated 5 mL of HCl and 30 mL of cold water. A white precipitate was formed, filtered off, and dried to give 0.18 g (93%) of compound 3 (Figure 1). Melting point (mp): 219–221°C,
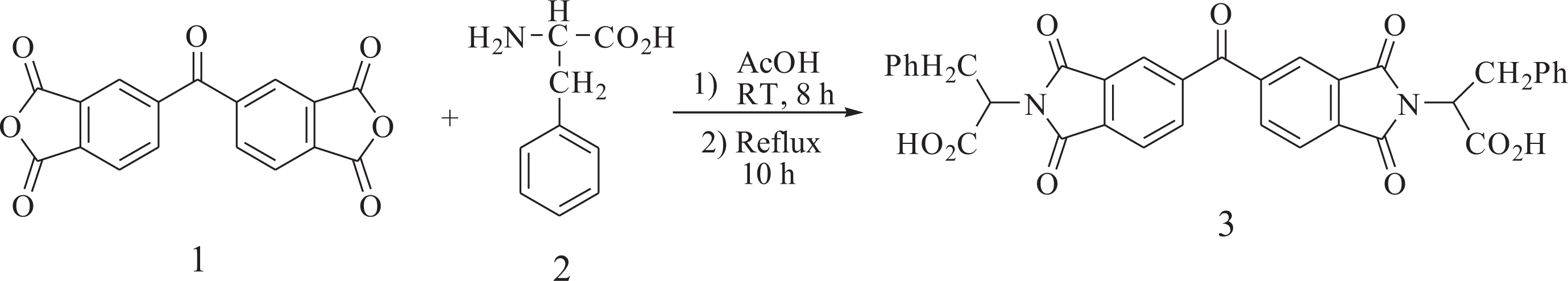
Synthesis of dicarboxylic acid 3.
Synthesis of diamine 10
Synthesis of 3,3′-((4-nitrophenyl)methylene)bis(1H-indole) (6)
A mixture of indole (1.55 g, 13.23 mmol) and 4-nitrobenzaldehyde (1.00 g, 6.62 mmol) in glacial acetic acid (5 mL) was refluxed for 6 h. The progress of the reaction was monitored by thin layer chromatography. After cooling, the reaction mixture was poured into 30 mL of water with constant stirring, producing a precipitate that was washed thoroughly with diethyl ether, collected by filtration, and dried at room temperature. Yield: 86%; mp: 225–227°C. FTIR (KBr, cm−1): 3427 (N–H stretch), 1507, 1331 (–NO2 stretch). 1H NMR (DMSO-d6, ppm): δ: 11.10 (s, 2 H, N–H), 8.21–6.89 (m, 14 H, Ar), 6.05 (s, 1 H); carbon-13 nuclear magnetic resonance (13C NMR) (DMSO-d6, ppm): δ: 154.00, 146.63, 137.47, 130.32, 124.73, 124.28, 127.24, 121.97, 119.78, 119.29, 117.55, 112.46, 40.35.
Synthesis of 4-(di(1H-indol-3-yl)methyl)aniline (7)
To a 50-mL two-necked flask, 0.200 g (0.544 mmol) of nitro compound 6, 0.01 g of 10% Pd/C, and 10 mL of ethanol were introduced. The suspension solution was refluxed, and 3 mL of hydrazine monohydrate was added dropwise. After the complete addition, the reaction was continued at reflux temperature for another 12 h. To the suspension, 10 mL of tetrahydrofurane (THF) was added to redissolve the precipitated product, and refluxing was continued for 1 h. The reaction mixture was filtered to remove the Pd/C and the filtrate was distilled to remove the solvent. The crude product recrystallized from ethanol and dried at 80°C under vacuum to give 0.143 g (yield: 78%) of compound 7. Mp: 230–232°C; FTIR (KBr, cm−1): 3413, 3386 and 3320 (N–H stretch). 1H NMR (DMSO-d6, ppm): δ: 10.69 (s, 2 H, NH), 7.38–6.51 (m, 14 H, Ar–H), 5.65 (s, 1 H, C–H), 4.83 (s, 2 H, NH2). 13C NMR (DMSO-d6, ppm): δ: 144.01, 137.10, 137.01, 129.59, 126.97, 123.40, 120.86, 119.75, 118.48, 118.08, 115.51, 110.72, 39.88.
Synthesis of N-(4-(di(1H-indol-3-yl)methyl)phenyl)-3,5-dinitrobenzamide (9)
To a 25-mL round-bottomed flask, 0.20 g (0.593 mmol) of the compound 7 in 5 mL of dry N, N-Dimethylacetamide (DMAc) was added. The reaction mixture was cooled in an ice water bath. To this mixture, 0.137 g (0.593 mmol) of 3,5-dinitrobenzoyl chloride (8) in 2 mL of DMAc was added dropwise. The mixture was stirred in an ice bath for 3 h, and, then, 0.083 mL of triethylamine was added. The mixture was stirred in an ice bath for 2 h and at room temperature overnight. The resulting mixture was poured into 25 mL/5 mL of cold water/concentrated HCl. The precipitate was collected by filtration and purified by recrystallization from DMF/H2O to afford dinitro compound 9 in 87% yield; mp: 330–332°C. FTIR (KBr, cm−1): 3426, 3264 (N–H stretch), 1650 (C=O stretch), 1546 and 1342 (–NO2 stretch). 1H NMR (DMSO-d6, ppm): δ: 10.85 (s, 2 H, N–H indole), 10.53 (s, 1 H, NH–C=O), 9.17–9.16 (d, 2 H, Ar–H), 9.01 (s, 1 H, Ar–H), 7.72–6.86 (Ar–H), 5.85 (s, 1 H, C–H).
Synthesis of 3,5-diamino-N-(4-(di(1H-indol-3-yl)methyl)phenyl)benzamide (10)
To a two-necked flask, 0.100 g (0.188 mmol) of the dinitro compound 9, 0.01 g of 10% Pd/C, and 10 mL of ethanol were introduced. The suspension solution was heated to reflux, and 3 mL of hydrazine monohydrate was added dropwise. After the complete addition, the reaction was continued at reflux temperature for another 12 h. To the suspension, 5 mL of THF was added to redissolve the precipitated product and refluxing was continued for 1 h. The mixture was filtered to remove the Pd/C and the filtrate was distilled to remove the solvent. The yield was 81%; mp: 263–265°C. FTIR (KBr, cm−1): 3461, 3436, 3361, 3326 (N–H stretch), 1652 (C=O stretch). 1H NMR (DMSO-d6, ppm): δ: 10.83 (s, 2 H, N–H indole), 9.89 (s, 1 H, N–H), 8.34–6.00 (Ar–H), 5.80 (s, 1 H), 4.00 (s, 4 H, N–H); 13C NMR (DMSO-d6, ppm): 167.92, 144.06, 138.71, 135.79, 130.11, 129.42, 128.62, 127.97, 122.63, 122.14, 118.67, 117.36, 110.56, 110.16, 100.74.
Polymer synthesis
A mixture of 0.20 g (3.24 × 10−4 mol) of dicarboxylic acid 3, 0.153 g (3.90 × 10−4 mol) of diamine 10, and 0.30 g (9.30 × 10−4 mol) of TBAB was ground until the powder was formed. After the mixture was completely ground, it was transferred into a 25-mL round-bottomed flask, and, then, 0.34 mL (1.30 × 10−3 mol) of TPP was added to the mixture, which was heated until a homogeneous solution was formed. Then, the solution was stirred for 12 h at 110°C, and the viscous solution was precipitated in 30 mL of methanol and the precipitated solid was filtered off and dried at 100°C for 6 h under vacuum to yield 0.31 g (87%) of the solid PAI. The inherent viscosity of the resulting PAI was obtained to be 0.52 dL g−1 and the specific rotation was measured (
Synthesis of GO
GO was synthesized according to modified Hummers technique. 34 Graphite powder (2.0 g), sodium nitrite (1.0 g), and potassium permanganate (6 g) were added to 60 mL of sulfuric acid (98%) and stirred in an ice bath for 90 min. Then, the reaction mixture was placed in a 40°C water bath and stirred for 2 h, followed by adding 150 mL of deionized water. Consequently, the temperature was elevated to 98°C and stirred for 30 min. After the temperature reduced to 60°C, 10 mL of H2O2 (30%) was added and stirred for another 2 h. The resulting product was filtered and washed with hydrochloric acid (5%) and deionized water until the pH of the washing water becomes neutral, and afterward, dried at 60°C for 12 h.
Synthesis of Fe3O4@GO
Fe3O4@GO was prepared by coprecipitation of FeCl3·6H2O and FeCl2·4H2O in the presence of GO. Forty milligrams of GO was dispersed in 40 mL of deionized water by ultrasonic irradiation for 30 min and 800 mg of FeCl3·6H2O and 300 mg of FeCl2·4H2O were separately dissolved in 50 mL of degassed deionized water under nitrogen atmosphere, and, then, this solution was added to the former suspension at room temperature. The temperature was increased to 80°C, and 10 mL of 30% ammonia solution was added to raise the pH to 10. Subsequently, being quickly stirred for 45 min, the reaction mixture was cooled to room temperature. The resulting Fe3O4@GO was washed with deionized water several times and dried at 60°C for 6 h.
Surface functionalization of Fe3O4@GO with APTES
Fe3O4@GO was placed in an oven at 110°C for 12 h to remove the adsorbed water. Fe3O4@GO was modified with APTES (H2N(CH2)3Si(OCH2CH3)3) as follows: 20 mg of Fe3O4@GO was dispersed in 20 mL of deionized water for 20 min using an ultrasonic bath. Then, 60 mL of ethanol was added and sonicated for 30 min. After the addition of 600 µL of APTES and stirring for 24 h at room temperature, the resulting product was collected via a magnet. The obtained product was washed with ethanol to remove additional APTES and dried at 60°C for 10 h.
Preparation of PAI/Fe3O4@GO nanocomposites
Different weight percentages of modified Fe3O4@GO (5, 10, and 15 wt%) were added to 0.2 g of PAI and the mixture was dispersed in 20 mL of absolute ethanol by ultrasound waves for 4 h. The obtained mixture was centrifuged. The resulting solid was dried at 80°C for 8 h.
Results and discussion
Monomer synthesis
The aromatic diamine containing a bulky pendent 4-(di(1H-indol-3-yl)methyl)phenyl unit, 3,5-diamino-N-(4-(di(1H-indol-3-yl)methyl)phenyl)benzamide (10) was synthesized through four-step synthetic route according to Figure 2. 3,3′-((4-Nitrophenyl)methylene)bis(1H-indole) (6) was prepared by electrophilic substitution reaction of indole and aldehyde in the presence of acetic acid as the activating agent. The catalytic hydrogenation of the nitro compound 6 to 4-(di(1H-indol-3-yl)methyl)aniline (7) was accomplished using hydrazine monohydrate and a catalytic amount of Pd/C. The dinitro compound 9 was synthesized by the amination reaction of compound 7 with 3,5-dinitrobenzoyl chloride (8), followed by hydrazine Pd/C-catalytic reduction. The purity of the intermediate compounds 6, 7, and 9 and the diamine monomer 10 was checked by thin-layer chromatography, which showed one spot in an ethylacetate/cyclohexane mixture (50:50). The chemical structures of the intermediate compounds 6, 7, and 9 and the diamine monomer 10 were proven with FTIR, 1H NMR, and 13C NMR spectroscopic techniques. The transformation of nitro to amino functionality could be monitored by the change of FTIR spectra. The nitro groups of compounds 6 and 9 gave two characteristic bands at around 1546 and 1342 cm−1 (–NO2 asymmetric and symmetric stretching). After reduction, the characteristic absorptions of the nitro group disappeared and the amino group showed the typical N–H stretching absorption pair in the region of 3461–3326 cm−1.
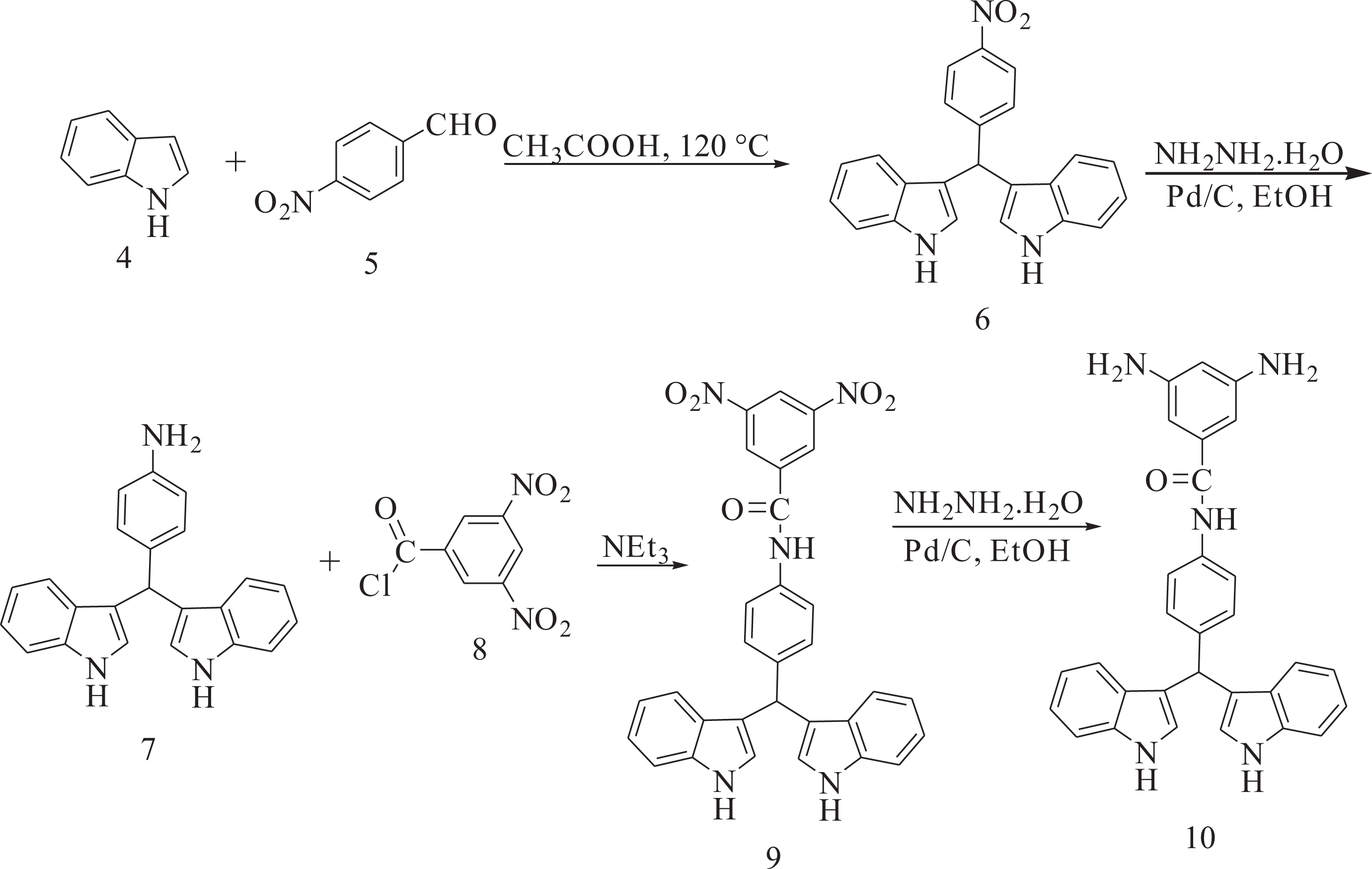
Synthesis of diamine 10.
Polymer synthesis
An optically active PAI was prepared via the polycondensation reaction of an equimolar mixture of dicarboxylic acid 3 with aromatic diamine 10 in glacial acetic acid (Figure 3). The inherent viscosity of the resulting PAI was 0.52 dL g−1 and the yield was 87%. The specific rotation of this polymer was
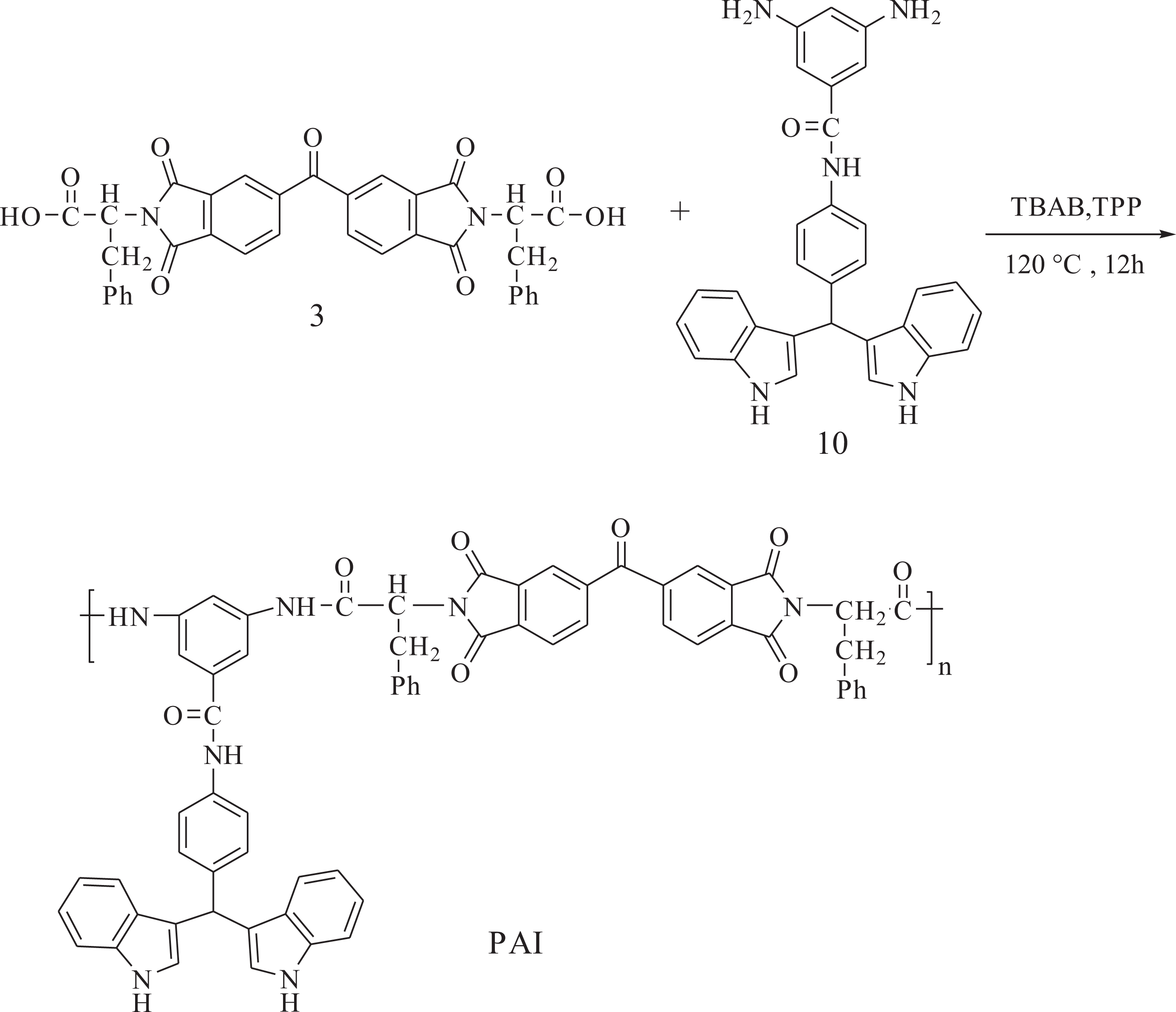
Preparation of PAI.
The structure of PAI was confirmed by FTIR and 1H NMR spectroscopy analysis. The FTIR spectrum of this polymer showed absorption bands around 3468 cm−1 (N–H), 1776 cm−1 (C=O asymmetric, imide), and 1721 cm−1 (C=O symmetric, imide). The presence of the imide heterocycle in this polymer was revealed by the absorption of 1386 and 727 cm−1, which belong to carbonyl bendings of imide (Figure 4(d)).
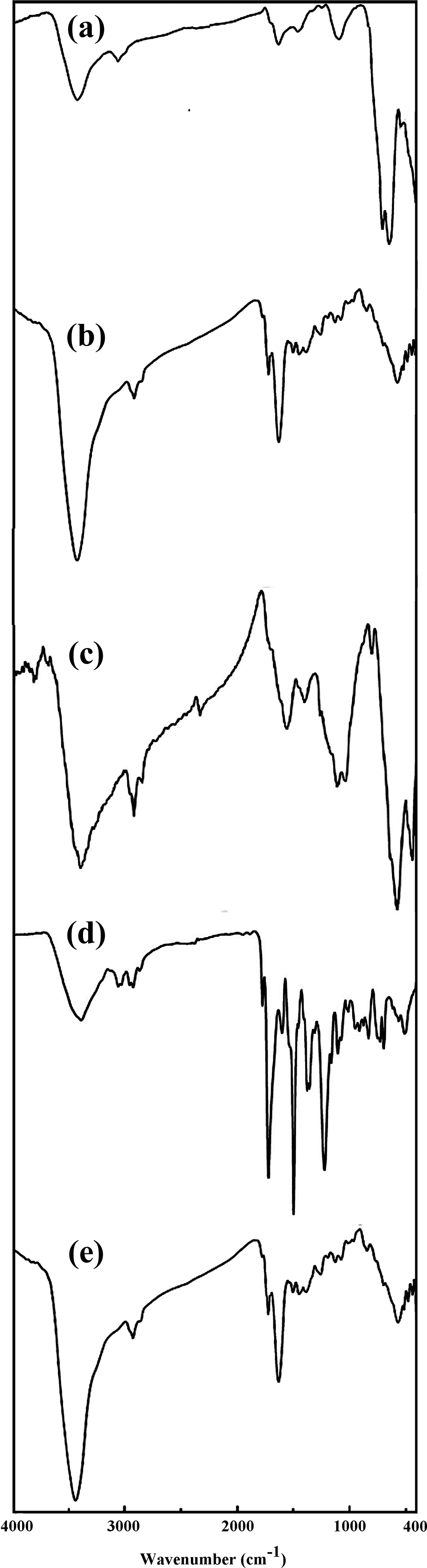
FTIR spectra of (a) Fe3O4, (b) Fe3O4@GO, (c) APTES-modified Fe3O4@GO, (d) PAI, and (e) PAI/Fe3O4@GO 10 wt%.
Preparation of PAI/Fe3O4@GO
The preparation process of PAI/Fe3O4@GO nanocomposites was shown in Figure 4. The PAI/Fe3O4@GO was prepared in a four-step process. At first, GO with oxygen functional groups was synthesized by oxidation of graphite powder according to a slightly modified Hummer’s method.32 The obtained GO was dispersed into water and treated with FeCl3.6H2O and FeCl2.4H2O to synthesize black Fe3O4@GO. Amine-functionalized Fe3O4@GO was synthesized through sonicating Fe3O4@GO with APTES. Surface-modifying Fe3O4@GO with an appropriate modifying agent can improve the compatibility of Fe3O4@GO with polymeric matrix, heightens efficiently the adhesion of polymeric matrix to Fe3O4@GO, and diminishes the aggregation of Fe3O4@GO in nanocomposites. Moreover, the addition of optically active PAI into the different weight percentages of Fe3O4@GO in ethanol produced magnetically separable PAI/Fe3O4@GO nanocomposites.
Characterization of nanocomposites
Features of the prepared nanocomposites were investigated with FTIR, XRD, SEM, VSM, and TGA. The FTIR spectra of Fe3O4 (a), Fe3O4@GO (b), APTES-modified Fe3O4@GO (c), PAI (d), and PAI/Fe3O4@GO 10 wt% (e) are shown in Figure 4. For Fe3O4@GO, the absorption band at 3415 cm−1 corresponds to stretching vibration of the surface O–H or the adsorbed water and the absorption peak at 1724 cm−1 is attributed to stretching vibration of C=O bands in carboxylic acid and carbonyl groups on the GO surface. The absorption band observed at 576 cm−1 corresponds to Fe–O stretching vibration. Additionally, the new absorption peaks observed at 2870–2948 cm−1 in the FTIR spectrum of modified Fe3O4@GO, which are attributed to C–H stretching vibration band of 3-amionopropyl moieties of APTES. From the spectral data, it can be deduced that the APTES has been grafted on Fe3O4@GO surface to generate an organic layer connecting with Fe3O4@GO. The FTIR spectrum of PAI/Fe3O4@GO revealed the same absorption peaks as the spectrum of Fe3O4@GO, but the two absorption peaks appeared at 1776 and 1723 cm−1, which are owing to the C=O asymmetric stretching vibration and C=O symmetric stretching vibration of imide units of the polymer. In addition, the peaks of 1386 and 729 cm−1 are attributed to the C–N stretching vibration and deformation vibration of imide rings. These results presented that PAI/Fe3O4@GO nanocomposites were successfully synthesized.
XRD patterns of the PAI (a), Fe3O4@GO (b), and PAI/Fe3O4@GO 10 wt% (c) (Figure 5) revealed that the diffraction peak at 2θ = 26° corresponded to the graphitic structure of GO. After preparation of Fe3O4@GO, seven diffraction peaks at 2θ values of 18.40°, 30.12°, 35.45°, 43.03°, 53.49°, 57.01°, and 62.60° were allocated to (111), (220), (311), (400), (422), (511), and (440) crystal planes of Fe3O4 nanoparticles, respectively. Besides, no other phases were appeared, which showed the purity of the product and outstanding efficiency of the preparation process for Fe3O4@GO. After modification by APTES and compositing with PAI (PAI/Fe3O4@GO), similar peaks can also be found. Moreover, the intensity of related peaks weakened, which was attributed to the surface coating of Fe3O4@GO.
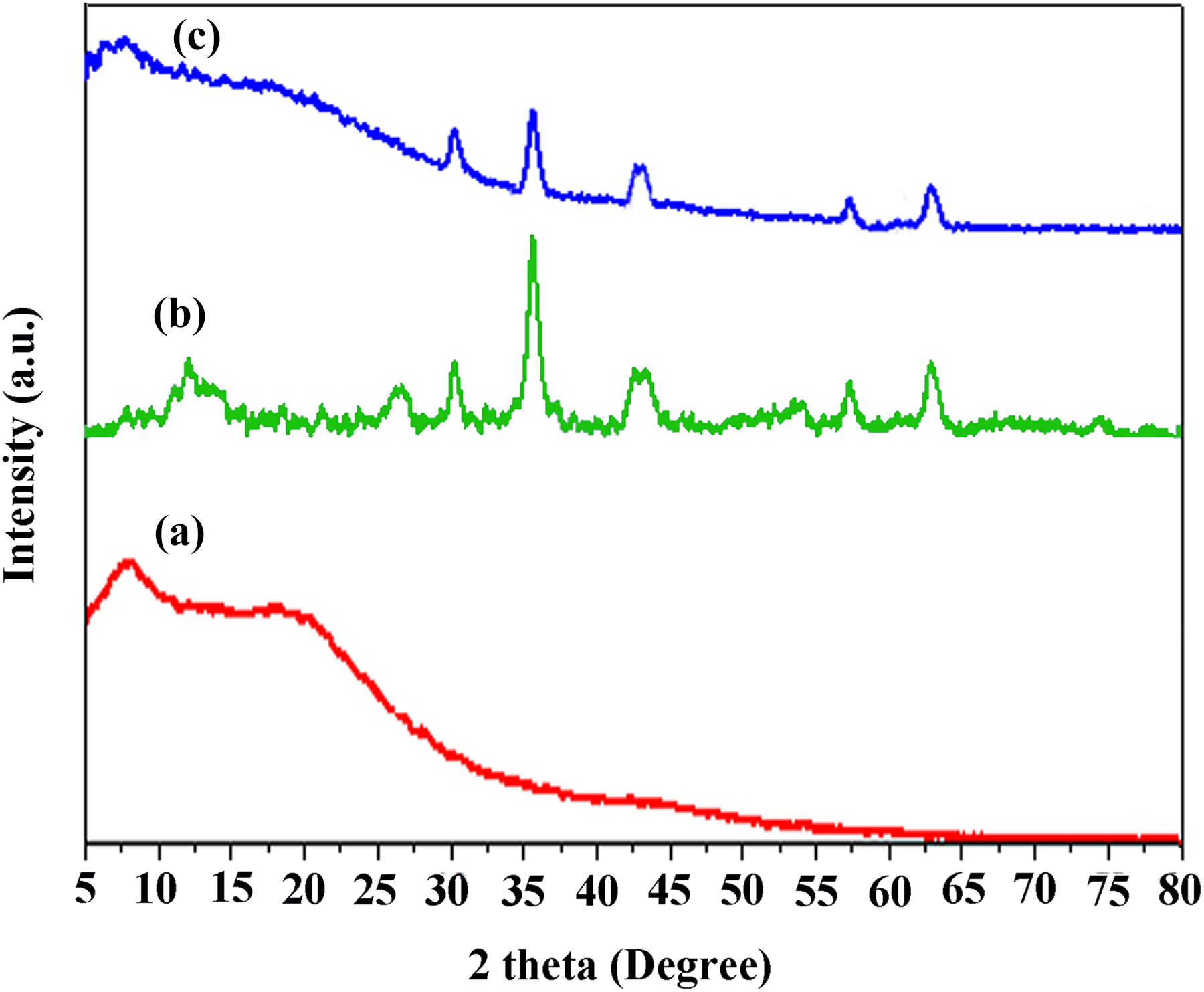
X-Ray diffraction patterns of (a) PAI, (b) Fe3O4@GO, and (c) PAI/Fe3O4@GO 10 wt%.
Figure 6 shows the SEM images of PAI (a), GO (b), Fe3O4@GO (c), and PAI/Fe3O4@GO 10 wt% (d). Clearly, the Fe3O4 nanoparticles uniformly decorated the surface of GO sheets (c). While after modification by APTES and compositing with polymer, the PAI/Fe3O4@GO nanocomposite became quite discrete from Fe3O4@GO (d). Although the dispersion state of magnetic nanoparticles was not altered, the interface of composite became ambiguous. From the SEM image, it can be found that the Fe3O4 nanoparticles were embedded into the GO sheets. This phenomenon was attributed to the fact that Fe3O4@GO was coated by organic layer owing to the modification by APTES and PAI.

SEM image of (a) PAI, (b) GO, (c) Fe3O4@GO, and (d) PAI/Fe3O4@GO 10 wt%.
The VSM curve of PAI/Fe3O4@GO 10 wt% at room temperature is shown in Figure 7. Hysteresis magnetic curve exhibits a behavior similar to that of superparamagnetic materials in terms of magnetization changes. The maximum saturation magnetization was 50 emu g−1. This value was lower than that of the bare Fe3O4 nanoparticles (81.9 emu g−1), which can be attributed to the presence of GO and PAI matrix. Hence, PAI/Fe3O4@GO nanocomposites can be successfully separated from the solution by a permanent magnet.
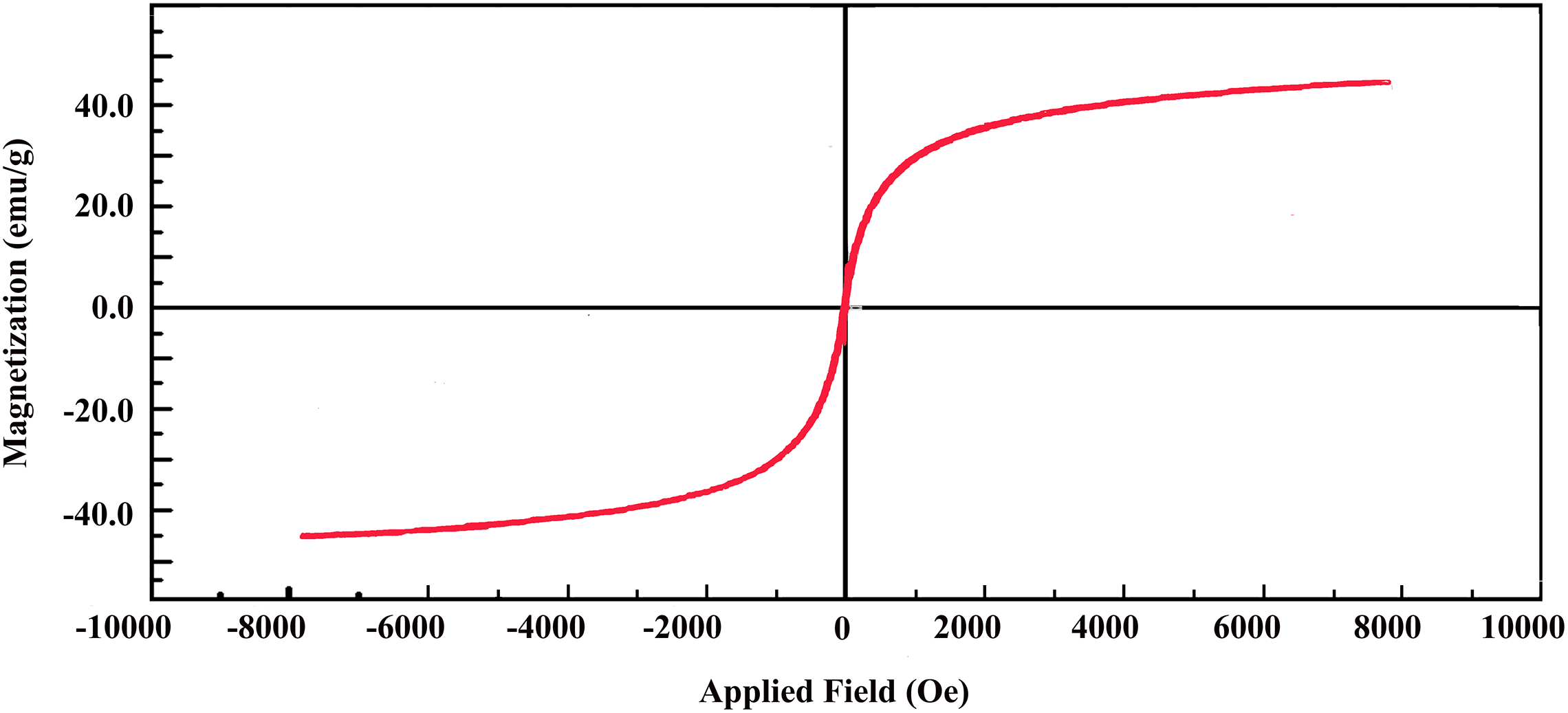
Magnetic hysteresis loops of magnetic PAI/Fe3O4@GO 10 wt%.
The thermal properties of pure PAI and PAI/Fe3O4@GO 10 wt% were studied using TGA at a heating rate of 10°C min−1 under nitrogen atmosphere. The TGA curves of pure PAI and nanocomposite 10 wt% were shown in Figure 8. The thermoanalysis data of these samples are summarized in Table 1. These studies show that the nanocomposites are thermally stable up to 500°C. The 10% weight loss temperatures of the pure PAI and nanocomposite under nitrogen atmosphere were recorded at 463°C and 549°C for PAI and nanocomposite 10 wt%, respectively. The amount of residue (char yield) of these nanocomposites under nitrogen atmosphere was more than 23% at 800°C. As shown in Figure 8, the thermal stability of nanocomposite is higher than pure PAI that is due to the good compatibility of modified Fe3O4@GO with polymer matrix. Fe3O4@GO has high thermal stability because of its larger surface area, so the incorporation Fe3O4@GO into polymer matrix can improve the thermal resistance of nanocomposites.
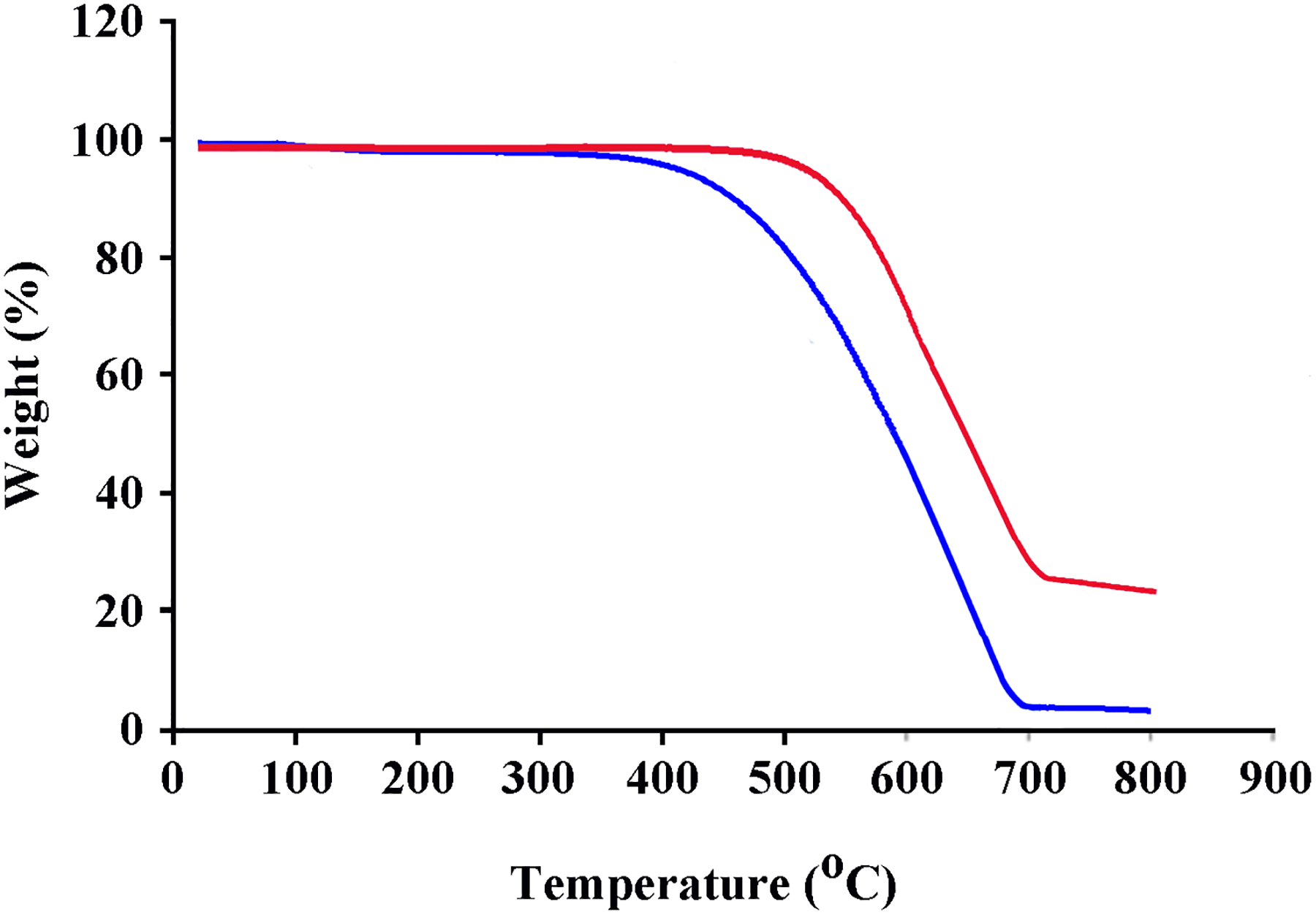
TGA thermograms of pure PAI and PAI/Fe3O4@GO 10 wt%.
Thermal properties of the pure PAI and PAI/Fe3O4@GO 10 wt%.
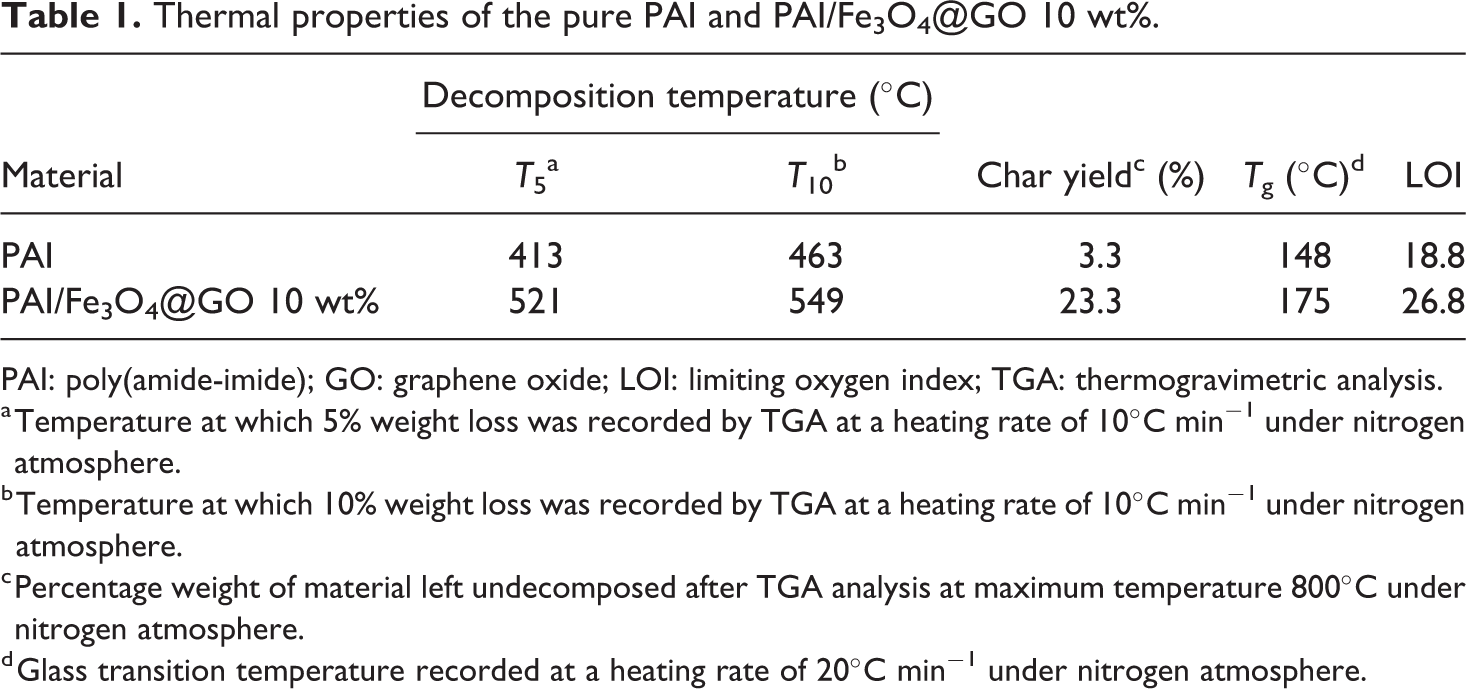
PAI: poly(amide-imide); GO: graphene oxide; LOI: limiting oxygen index; TGA: thermogravimetric analysis.
a Temperature at which 5% weight loss was recorded by TGA at a heating rate of 10°C min−1 under nitrogen atmosphere.
b Temperature at which 10% weight loss was recorded by TGA at a heating rate of 10°C min−1 under nitrogen atmosphere.
c Percentage weight of material left undecomposed after TGA analysis at maximum temperature 800°C under nitrogen atmosphere.
d Glass transition temperature recorded at a heating rate of 20°C min−1 under nitrogen atmosphere.
Char yield can be applied as a decisive factor for estimating limiting oxygen index (LOI) of the polymers based on Van Krevelen and Hoftyzer equation 35 :
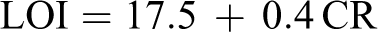
where CR is char yield.
The LOI values calculated and derived for nanocomposites from their char yield were higher than 26. On the basis of LOI values, these nanocomposites can be classified as self-extinguishing materials.
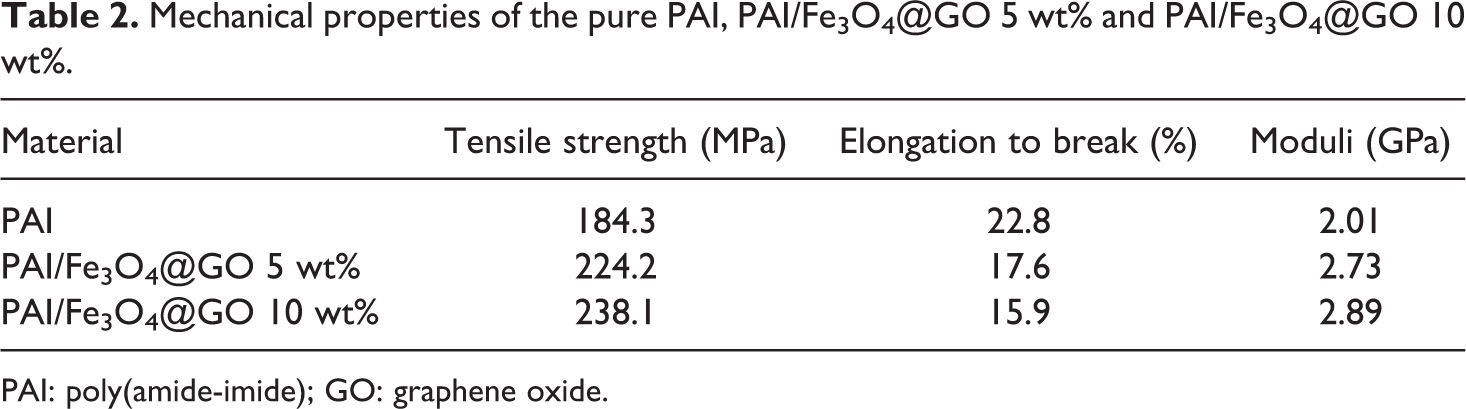
The mechanical properties of the nanocomposites at room temperature are collected in Table 2. Compared with the pure PAI film, with the increase of the Fe3O4@GO content, the tensile strength and modulus of the nanocomposites increase due to the reinforcing effect of Fe3O4@GO.
Mechanical properties of the pure PAI, PAI/Fe3O4@GO 5 wt% and PAI/Fe3O4@GO 10 wt%.
PAI: poly(amide-imide); GO: graphene oxide.
Adsorption of Hg2+ ion
Five milligrams of PAI/Fe3O4@GO 10 wt% was added to 25 mL of metal ion solution with the initial metal ion concentration of 10 mg L−1 and stirred for 3 days at room temperature. After centrifuge separation, the amount of adsorbed ion was calculated. qe, R, and remaining ion concentration of PAI/Fe3O4@GO 10 wt% obtained were 28.11 mg g−1, 63.21%, and 3.83 ppm, respectively.
Conclusion
For the first time, the novel well-defined superparamagnetic PAI/Fe3O4@GO tricomponent nanocomposites were prepared by in situ growth of Fe3O4 on GO, modification of Fe3O4@GO with APTES as a surface modifier, and compositing with the chiral PAI in the presence of ultrasound waves. It showed excellent paramagnetic, mechanical, and thermal properties. The SEM analysis exhibited dispersion of Fe3O4@GO with average particle size of about 30 nm in the polymer matrix. The TGA curves presented that the addition of Fe3O4@GO into the PAI could enhance the thermal stability of polymers. Additionally, the results demonstrated that these nanocomposites show an excellent adsorption capacity toward Hg2+ ion from aqueous solution.