
Abstract
Kinetic rigidity of several polymers such as 2,6-bis(3-aminophenoxy)benzonitrile/4,4′oxydiphthalic anhydride (β-CN)APB/ODPA, poly[(2,2-dimethyl-1,3-dioxolan-4-yl)methyl acrylate)] (PACGA), and diglycidyl ether of bisphenol A (DEGEBA) was studied. Rigidity parameter D, Vogel’s temperature T0, and the activation energy Uα (Tg) for the glass transition were evaluated through Vogel’s model along with relaxation data using “nonlinear” regression of Arrhenius function. The existence of certain functional groups within the structure, such as the aromatic rings, gives high level of kinetic rigidity to the structure as is the case of (β-CN)APB/ODPA and DEGEBA, while the aliphatic groups confer flexibility, as in PACGA.
Introduction
Nowadays, polymers have substituted certain materials that a few years ago were considered essential for industrial and everyday use. The need for new materials with better properties and easy to manufacture gave, as a result, a significant progress in research on new polymers. The results of such research are now used in different areas of the industry such as aerospace, automotive, and electronics, among others. When new polymeric structures are proposed, it is of primary interest to know the dielectric and dynamic mechanical response of the polymer chains to electric and mechanical force fields due to the dependence of physical performance on polymer relaxation properties. 1 Generally, in polymeric materials, the mechanical and dielectric responses present one or more relatively weak absorptions in the glassy state, followed by a broad relaxation associated with the glass to rubber transition. The glass to rubber relaxation is assumed to be the result of segmental or micro-Brownian motions, and frequency or time relaxation distributions are the parameters used to determine the performance of the material in force fields. For example, there are different characteristics that determine the final use of a given material; for instance, if a polymer in the glassy state suffers low deformations, it will show elastic behavior; however, if the same material suffers high deformations, it will show viscous behavior. The different mechanical properties of polymers not only depend on the type of stress and deformation fields that are applied and the application time, 2 but it will also depend on polymer characteristics such as the glass transition temperature (Tg), crystallinity, density, chain packing, and morphology among other factors. 3 Other characteristics of polymeric materials are the high level of dependence that its mechanical properties present regarding the molecular weight, up to a certain level, as well as the Tg values since Tg is linked to molecular chain dynamics, and it is the result of the coordinated motions of its atoms.
The study of the polymeric chain dynamics based on its length and scale can be classified as global or local. The first ones refer to large motions within the chains and are the most important for our study; however, the study of local dynamics is important since such dynamics have macroscopic consequences over the final properties of the materials.
4,5
Both the local and the global motions strongly depend on the chemical structure of the material, and, therefore, they depend on the existing functional groups. That is the reason why making changes in a functional group within the structure of a polymer, the thermodynamic and chain packing properties can be affected, and this modifies some macroscopic properties, as has been shown in some studies, where structural substitutions systematically modify the thermal properties such as Tg due to a disruption of symmetry or introduction of pendant group that expand mobility chains.
6
–15
Moreover, relaxation properties are also related to molecular structure, which in turn determines that a certain material presents suitable mechanical properties. In the glassy state, some polymers are characterized by plastic properties, whereas other polymers show well-pronounced fragile properties under the action of high stresses. The first group involves structurally mobile polymers, while the second group includes structurally fragile polymers. One way to determine whether a certain polymer has suitable plastic properties or fragile properties would be found by characterizing its structural rigidity. This structural rigidity is an intrinsic property of polymeric materials, and it is formed by two contributions.
16
The kinetic rigidity (D), characterized by the mobility of chain units that is determined by the potential barriers for rotation, and the thermodynamic or conformational rigidity (Cn), which is characterized by the dimension of polymer chains
There are some parameters that are related to kinetic rigidity such as fractional free volume (FFV), which is defined as the average effective space between chains; Vogel’s temperature (T0), which determines the temperature at which the material present the higher levels of rigidity due to the hindrance of chains to relax; and Tg, which is defined as the temperature at which the material goes from a glassy state to a rubbery one.
It is well-known that mechanical properties of a polymer depend on temperature; however, it is also known that if a mechanical sinusoidal stress is applied to the material, the mechanical response will also depend on the frequency of excitation. According to this, when the elastic modulus is used as a measure of structural rigidity, its value is determined only for a certain absolute value of temperature. Another parameter that is used as a measure of structural rigidity is the activation energy at the glass transition because it measures the energy required to carry out the relaxation processes leading to Tg. Given this problem, a parameter to globally measure structural rigidity (instead of locally) is required to validate this measure for a wide interval of temperatures.
The relaxation phenomenon, whether depending on temperature or frequency, will show a T0 temperature (Vogel’s temperature) at which the material will present its maximum rigidity due to the lack of Brownian motions that allow relaxation, as well as an interval of temperatures at which there will be chains of short-range motion, where the relaxations of chains are concentrated, and as a consequence will correspond to the interval of flexibility of the material, with Tg being the temperature where the maximum flexibility is present. The same occurs if we use the relaxation spectrum at a constant temperature as a function of frequency; here, due to the time–temperature superposition principle, 3 the frequency of maximum flexibility can be related to Tg, while the frequency of maximum rigidity can be related to T0. According to the aforementioned, it is possible to consider the interval of temperatures at which the relaxation occurs and in which there is mobility of chains; as a consequence, it is possible to globally measure the structural rigidity, because the size of the interval (Tg–T0) represents a measure of flexibility. If a certain material shows a very short interval of temperatures at which relaxations are present, it is said that the material is rigid (fragile polymer), if the contrary takes place, then the material is said to be flexible (mobile polymer). 19 –21
This work is intended to analyze kinetic structural rigidity in a set of amorphous polymers of technological interest, using their dynamic–mechanical or dielectric spectra of relaxation. It aims at measuring the kinetic rigidity through a dimensionless parameter using Vogel’s model, 19 –21 which is valid for amorphous polymers, to eventually try to relate the effect that polymer structure has over the properties of structural rigidity. Particularly, the parameter of structural kinetic rigidity (D) is studied to know the temperature interval at which the material undergoes relaxation process or chain mobility. To do so, published results related to dielectric relaxation (DRA) spectra at constant frequency depending on the temperature or at constant temperature measured at different frequencies in three different materials are utilized: polyimide 2,6-bis(3-aminophenoxy)benzonitrile/4,4′oxidiphthalic anhydride (β-CN)APB/ODPA, an acrylic polymer poly[(2,2-dimethyl-1,3-dioxolan-4-yl) methyl acrylate)] (PACGA), and an aromatic polymer diglycidyl ether of bisphenol A (DEGEBA).
Theory
Shift of glass transition depending on the frequency
Based on an analysis of the dependence with temperature and frequency of polymers dynamic–mechanical behavior, there is a relationship between these two variables. The phenomenon that relates this dependence is known as time–temperature superposition principle, 3 which states that an increment in temperature has the same effect as if there was an increment in the frequency or a decrease in time over the measured viscoelastic behavior. Figure 1 shows this effect, and as can be observed, the dynamic–mechanical relaxation behavior dependence on temperature (constant frequency) shows a shift of glass transition at higher temperatures as a result of a change in excitation at higher frequencies.
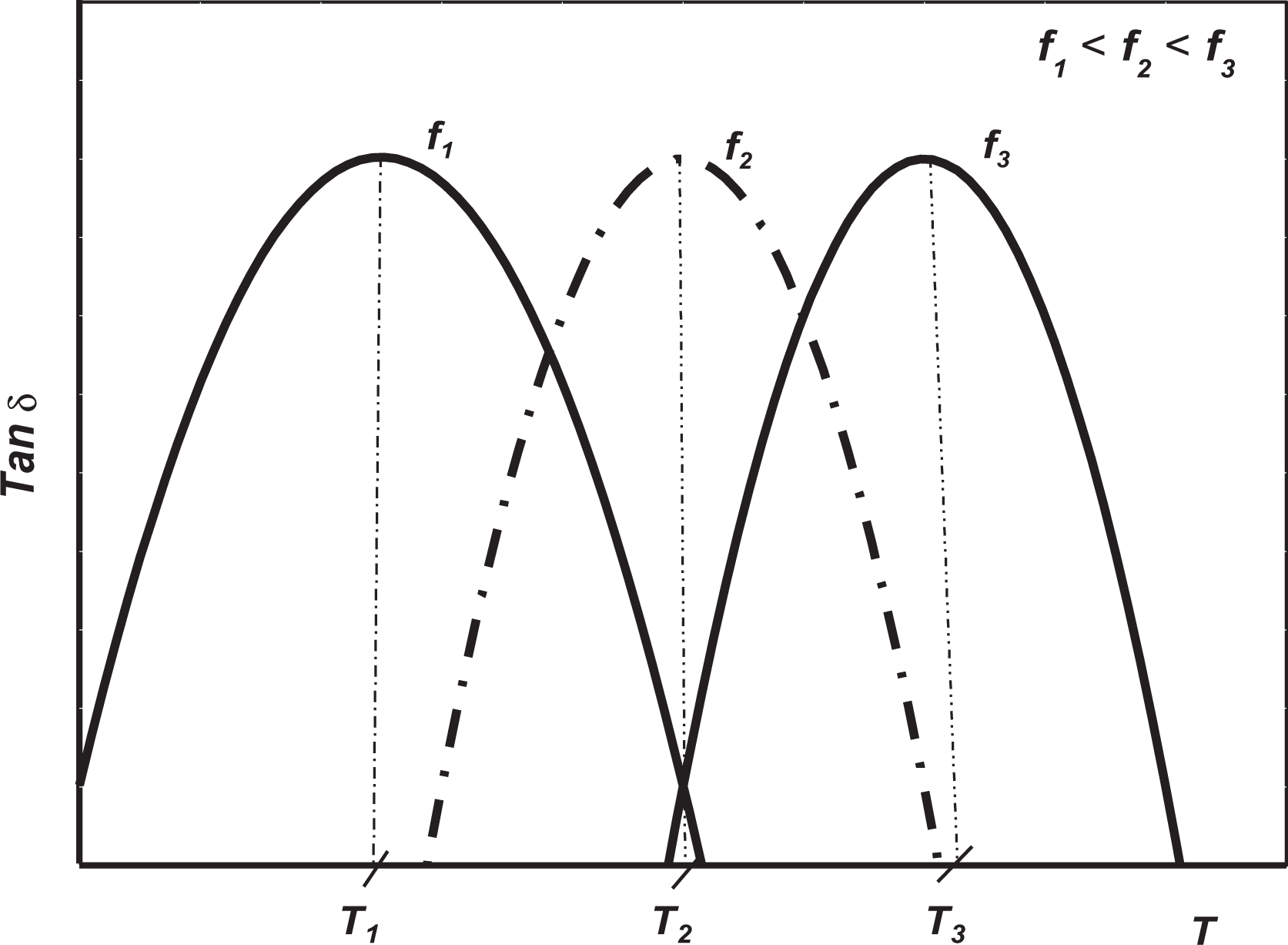
Graphic of a shift in glass transition depending on frequency.
This behavior can be modeled as an Arrhenius-type equation, 3,20 –22 with a correlation that defines a dependence of the relaxation times for the glass transition as a function of temperature as shown in equation (1)

where the relaxation time can be defined by the frequency of excitation as follows

In equation (1), Uα represents the activation energy of the relaxation process, Bα represents the relaxation time at infinite temperature, and k = 8.29 J/(mol K) is Boltzmann’s constant.
In the case of amorphous polymers, they can be modeled at the α-transition process using Vogel’s method for the activation energy through the following equation 20,21

where U∞ represents the lower limiting value of the activation energy that is obtained when T →
Structural rigidity parameter “D”
Using the last model, it is possible to obtain a parameter to measure the interval of temperatures where there is molecular flexibility. By substituting equation (3) in equation (1), the transition α just in the glass transition (T = Tg) is obtained using the following equation

The validity of equation (4) is when it is defined
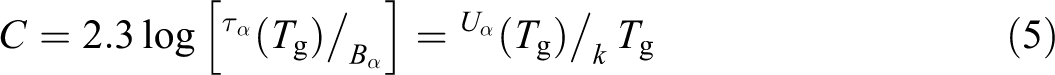
and
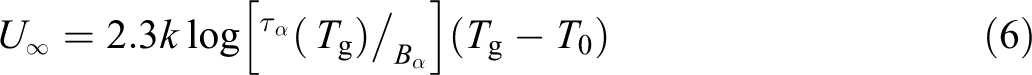
In this way

with the activation energy

There is possibility to identify three important characteristics in D: Parameter D identifies the minimum energy, U∞ (occurring at T → ∞), required by a polymeric system to carry out the relaxation process. Parameter D can go from 0 to The lower the D is, the higher the material’s fragility is.
In equation (7), it is possible to observe that there is a linear dependence between the dimensionless parameter D with the temperatures ratio (Tg/T0). This ratio of temperatures can be used as a measure of the interval where there is structural mobility in amorphous polymers because Tg is the temperature of maximum flexibility and T0 is the temperature of maximum rigidity. Then, the higher the D is, the lower is the rigidity of the amorphous polymer since the interval of temperatures at which the segmental mobility occurs increases. Figure 2 describes this behavior. Moreover, slope C of the linear behavior identifies the energy required at Tg by the system to carry out the α-relaxation process.
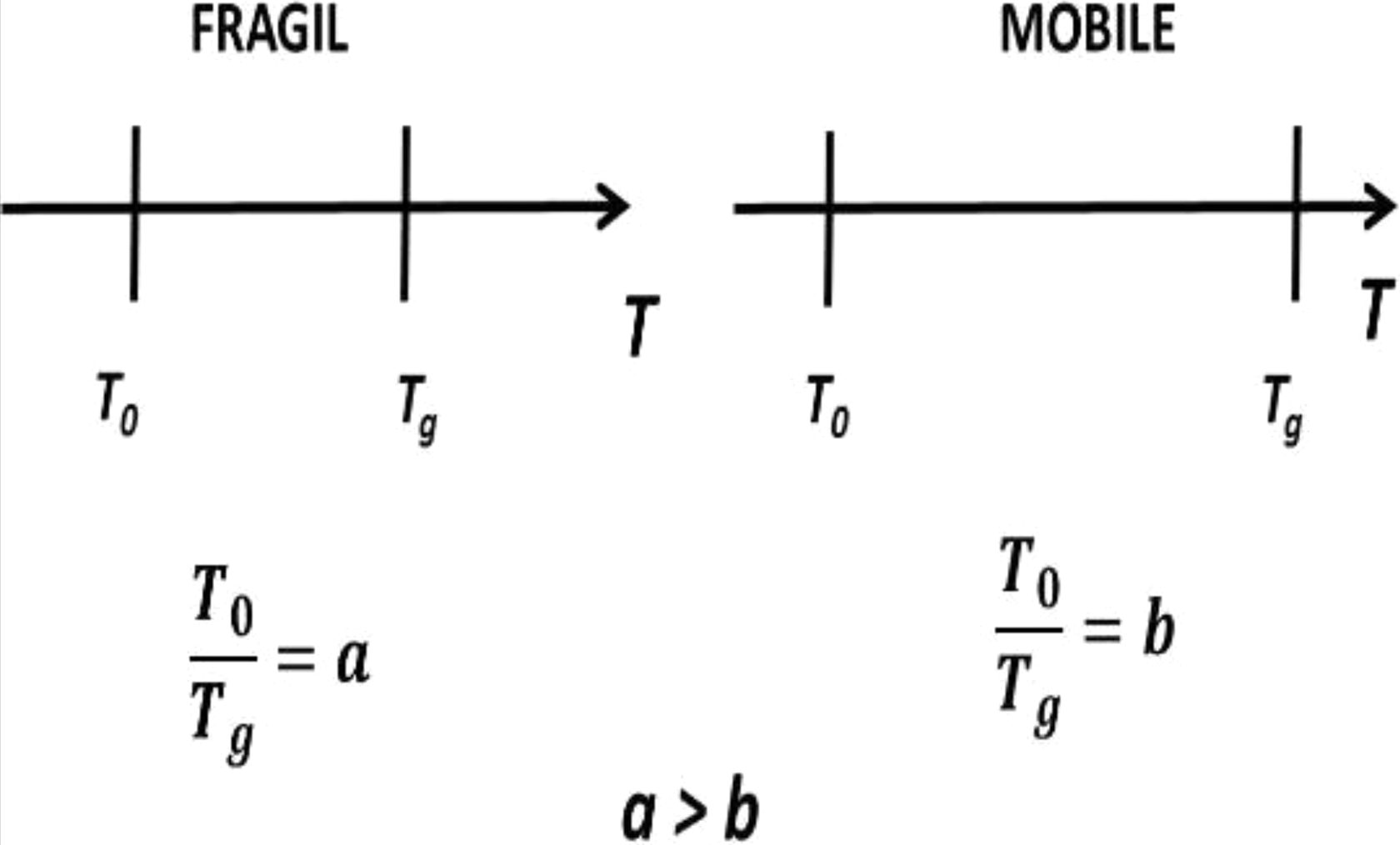
Graphic of structural rigidity parameter as a function of Tg and T0.
Using the shift of temperature that occurs at the α-transition according to the frequency of dielectric excitation that can be observed in the storage modulus E′, loss modulus E″, or in tan δ, it is possible to characterize Vogel’s parameters through the Arrhenius function (equation (5)).
To do that, the measurement of the DRA spectra of a polymer as a function of temperature at constant frequency is required; such measurement should be done using at least four different frequencies to obtain the shift on α-transition and the maximum of the transition, in either the loss modulus (E″) or damping factor (tan δ) for each frequency. However, based on the relationship of the time–temperature superposition principle, it is also possible to use the DRA spectra as a function of frequency at a constant temperature. Since the frequency is related to the relaxation time τα according to equation (2), it is possible to obtain a graphic of τα as a function of temperature T where the maximum of α-transition takes place, in other words at Tg. Finally, a nonlinear regression on this graphic using equation (5) permits to obtain parameters D, Bα, and T0, as has been reported before. 19
On the other hand, due to the universal character of the Arrhenius equation for the α-relaxation behavior in polymers, the pre-exponential coefficient Bα has been proposed in other works as an invariant for systems of flexible chains with a value of 5 × 10–12 s (±20%). 3 –5
In Table 1, it is possible to observe Vogel’s parameters for several structures that have been characterized in other works through DRA. 2 Regarding the materials presented in this table, it can be seen that the obtained structure is polydimethylsiloxane with higher levels of kinetic rigidity having the value of D = 21.8, indicating wider segmental mobility for the interval of temperatures Tg – T0 when compared with the rest of the polymers in the same table. On the other hand, the structure with lower flexibility is the one that corresponds to polyethylene terephthalate (PET), with D = 4.0 being its parameter of kinetic rigidity, showing an indication of narrow segmental mobility and is at the Tg midpoint.
Vogel’s parameters in some amorphous polymers. 3
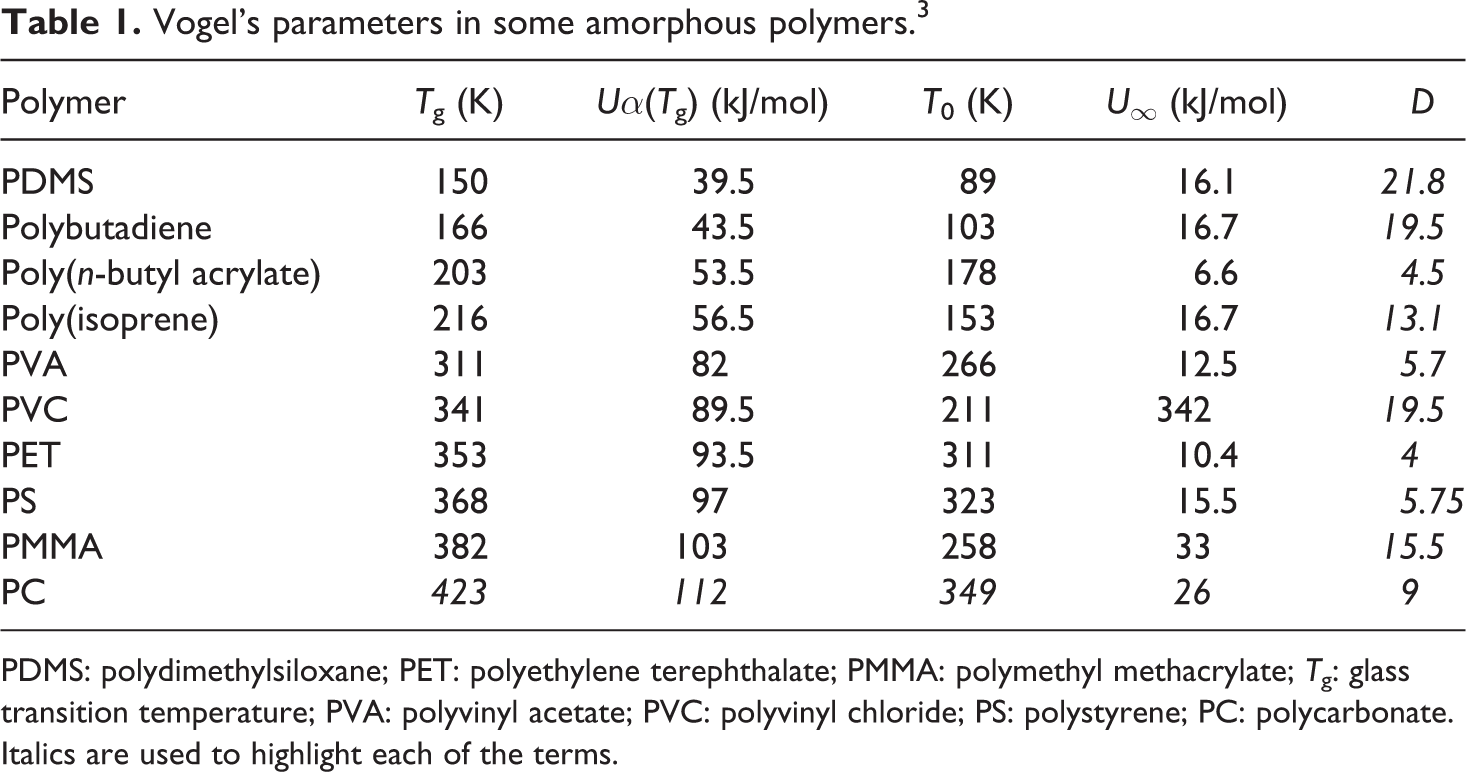
PDMS: polydimethylsiloxane; PET: polyethylene terephthalate; PMMA: polymethyl methacrylate; Tg: glass transition temperature; PVA: polyvinyl acetate; PVC: polyvinyl chloride; PS: polystyrene; PC: polycarbonate.
Italics are used to highlight each of the terms.
Results and discussion
Case materials and their relaxation properties at the glass transition
The polyimide (β-CN)APB/ODPA
Some years ago, researchers at NASA synthesized the polyimide called (β-CN)APB/ODPA, which was obtained using 2,6-bis(3-aminophenoxy) benzonitrile ((β-CN)APB) and 4,4′oxydiphthalic anhydride (ODPA), having N,N-dimethylacetamide as the reaction medium. 22 This reaction takes place at room temperature under nitrogen atmosphere to obtain polyamic acid, and later a hydrolysis process is applied to obtain the final structure. The chemical structure and the synthesis route for this material are shown in Figure 3.
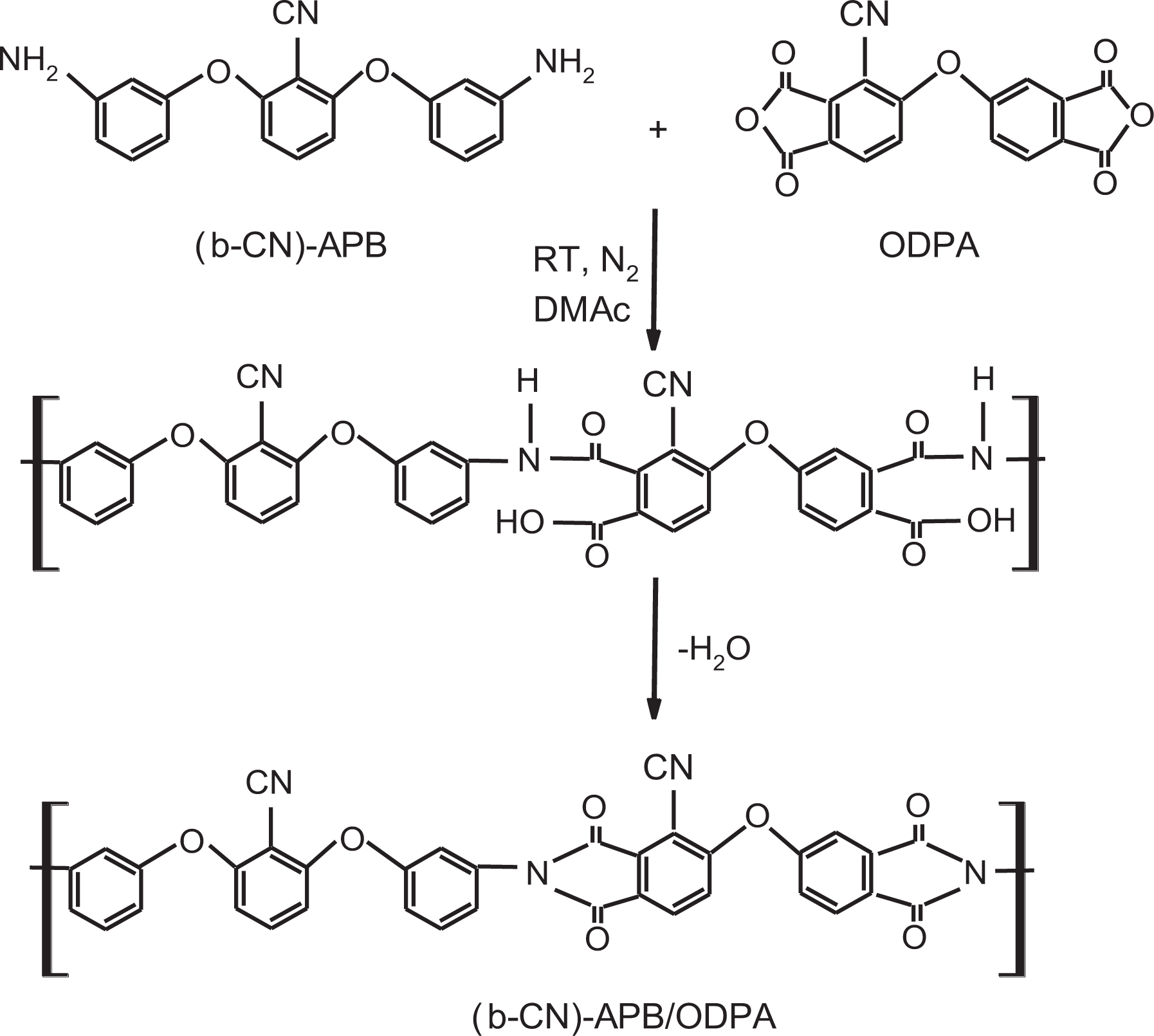
Synthesis and structure of (β-CN)APB/ODPA. 22
As can be observed in Figure 3, the chemical structure of this material is totally aromatic, which leads us to believe that its kinetic rigidity should correspond to one of the materials with a low D parameter; in other words, a material with a low level of flexibility because different research studies indicate that the aromatic rings confer rigidity to polymer structures. 16 It is worth mentioning that the characterization reported for this material 22 indicates that this is an amorphous material; therefore, Vogel’s model for the glass transition can be applied.
As part of this report, the results of dielectric relaxation analysis in this structure at four different frequencies (10 Hz, 100 Hz, 1 kHz, and 10 kHz) were presented too, in which the dielectric modulus was evaluated as a function of temperature. 22 A shift in glass transition as a function of frequency was observed, in agreement with the time–temperature superposition principle. In Table 2, we can observe the effect of frequency over the maximum of the α-transition (equivalent to the Tg); that is, for this case, it was chosen from the storage modulus as the medium point of the interval at temperatures where the relaxation takes place. The behavior presented was at 10 Hz, the Tg was 429.21 K; at 100 Hz, it changed to 434.92 K; at 1000 Hz was 446.35 K; and finally at 10,000 Hz, the transition was visible at 452.07 K.
Experimental data in the α-relaxation process of (β-CN)APB/ODPA.
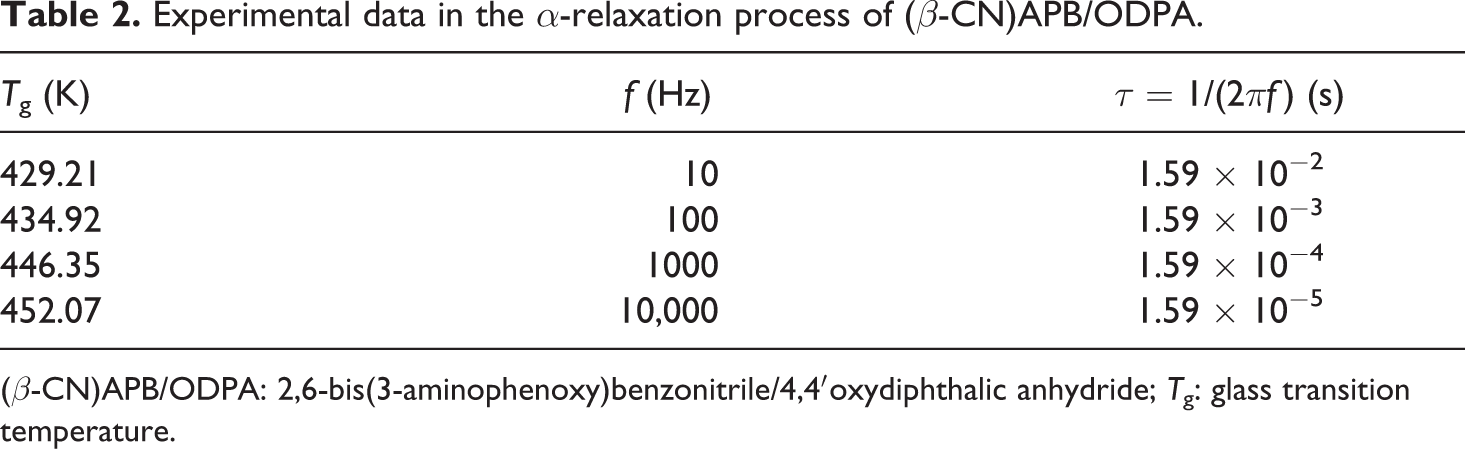
(β-CN)APB/ODPA: 2,6-bis(3-aminophenoxy)benzonitrile/4,4′oxydiphthalic anhydride; Tg: glass transition temperature.
Figure 4 is a graphic representing the relaxation times (τα) as a function of the temperature (T) where the α-transition appears in the polyimide. The black points represent the experimental data regarding the relaxation time as a function of Tg. Due to the fact that the Vogel’s model fulfills this behavior, we will use a “nonlinear” regression, based on equation (5), which represents this model to obtain the structural rigidity. This “nonlinear” regression corresponding to the relaxation times as a function of temperature in the Vogel’s model for the structure is also present in the experimental data of Figure 4.
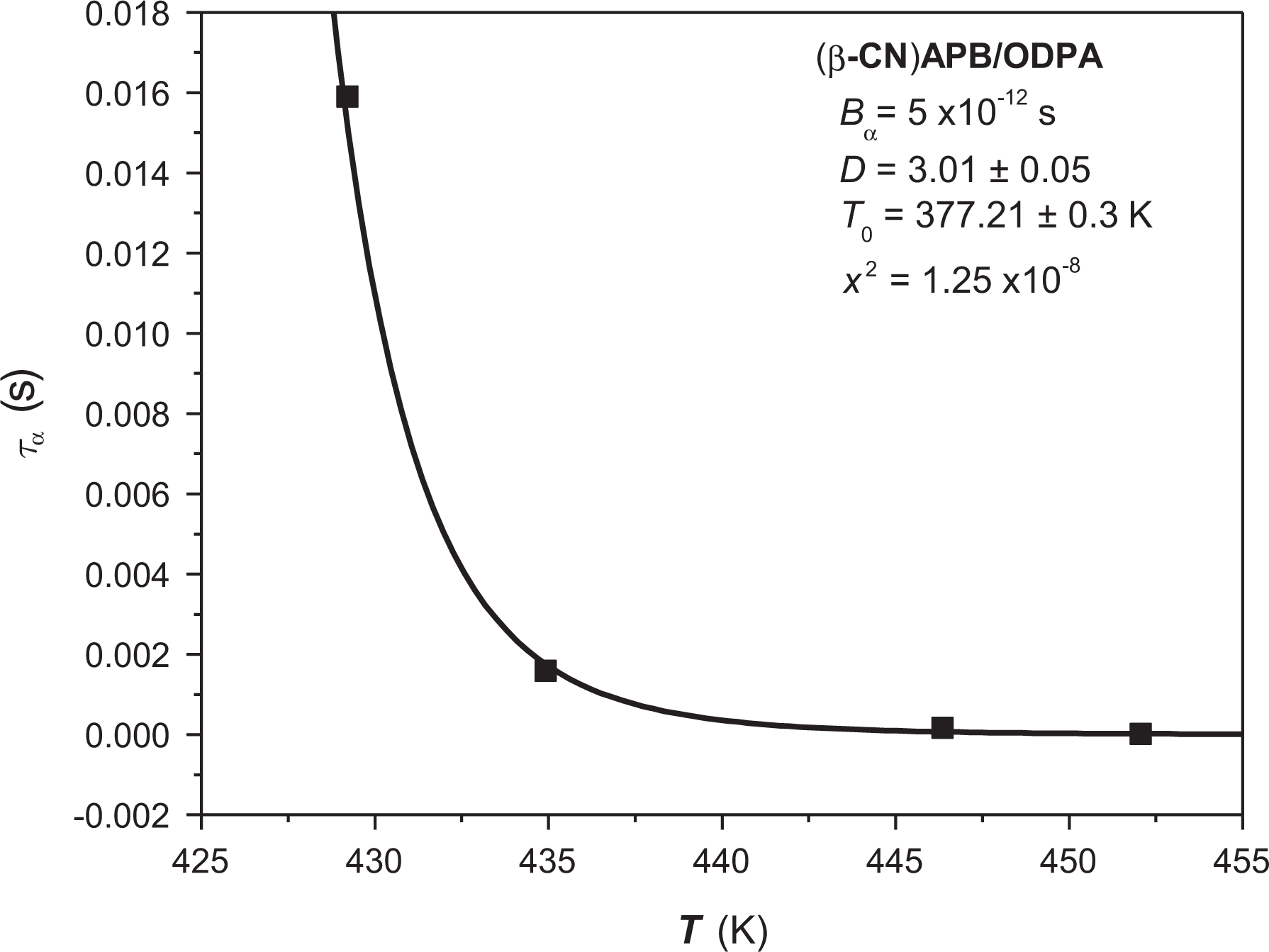
“Nonlinear” regression for α-relaxation of (β-CN)APB/ODPA.
The regression parameters for this case were the Vogel’s temperature or temperature of maximum rigidity T0 and the structural rigidity parameter D, due to the fact that the time of relaxation at infinite temperature (Bα) was 5 × 10−12 s, according to the results obtained in other works where it is suggested that this value is an invariant for polymeric systems. 19 –21
It is possible to observe that the regression presents low levels of dispersion, which is confirmed by the small value of the mean square deviation χ2 that was equivalent to 1.59 × 10−7. Such mean square deviation is obtained by dividing the difference of squares of the experimental relaxation time τα = 1/2Πf among the results obtained in the adjustment. The Vogel’s parameters obtained are Bα = 5 × 10−12 s, D = 3.01 ± 0.05, and T0 = 377.21 ± 0.3 K. It should also be considered that Tg reported in studies of differential scanning calorimetry (DSC) is equivalent to 415 K. 22
As it was explained before, the first parameter indicates the relaxation time at infinite temperature, the second one is a measure of the inverse of the rigidity and can be used to compare the effect of the structure over the structural mobility, and the last one indicates that below T0 the structure is completely rigid. Once stated the aforementioned, it is evident that the polyimide (β-CN)APB/ODPA presents high kinetic rigidity in comparison to the data in Table 1, which contains characteristic data regarding kinetic rigidity of some polymers.
As for the analysis of the functional groups that are present in this structure, it has been proposed that its high rigidity can be a consequence of the presence of the aromatic rings as stated in other works in which the effect of the aromaticity over the kinetic rigidity has been the subject of research, and it was established that the aromatic rings are responsible for increasing the structural rigidity. 16
On the other hand, due to the fact that the Vogel’s temperature for this structure is equivalent to 378.51 K, the use of this material in processes that involve temperatures lower to 105.51°C is not recommended because plastic properties at this temperature diminish as a consequence of the lack of mobility on its structure. 16
Poly[(2,2-dimethyl-1, 3-dioxolan-4-yl) methyl acrylate]
In the last few years, the behavior of the aliphatic groups has been studied during the relaxation process of acrylic structures. 23,24 It is known that the presence of aliphatic functional groups in certain polymers gives flexibility to the molecular structure, 16,24 as it has been studied in polymethyl methacrylate (PMMA) for which high flexibility (D = 15.5) is a result of the methyl groups (CH3) present in this structure. 16
It is worth mentioning that PACGA is an acrylic whose structural difference with PMMA lays on the presence with respect to PMMA structure of the aliphatic ramification at the methyl, whereas in the PACGA structure that derives from polymethyl acrylate, the dioxacyclopentane ring is added as lateral group and the aliphatic ramification of PMMA is no longer present. Figure 5 shows a schematic of the chemical structures of PACGA and PMMA, in such schematic, it is possible to observe the structural differences between both structures.
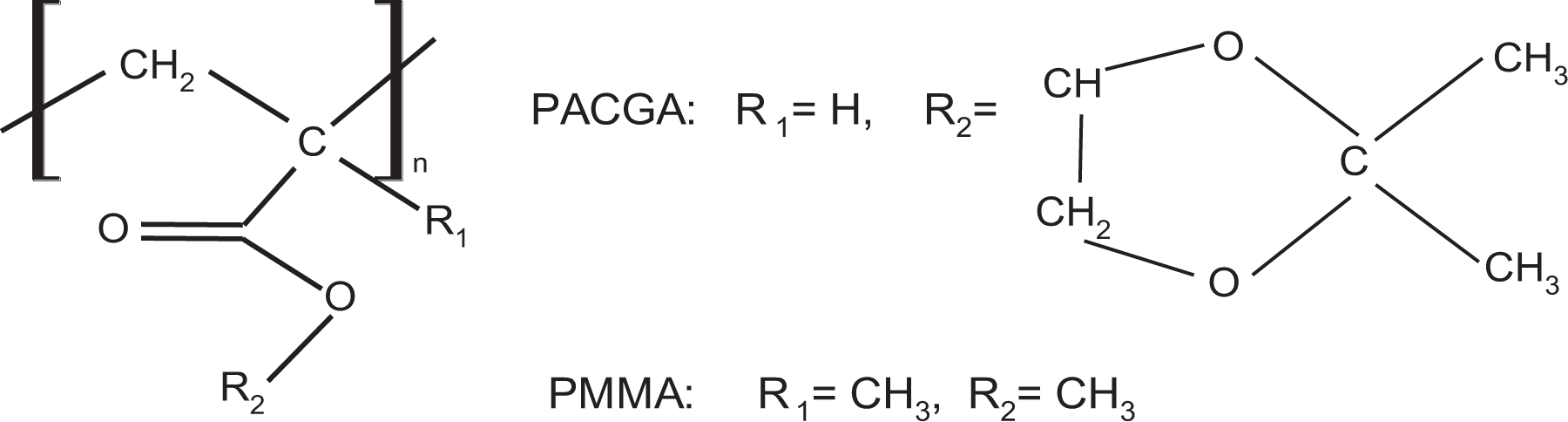
Structure of the dioxacyclopentane ring added as a lateral group at the acrylic structure PACGA. 23
According to that analysis, the shift of α-transition was observed as a function of frequency. In this case, the transition temperature Tg was chosen as the one that corresponds to the maximum of the main transition in the loss modulus. The shift of the α-transition for this structure is tabulated in Table 3.
Experimental data of the α-relaxation process of PACGA. 23

PACGA: poly[(2,2-dimethyl-1,3-dioxolan-4-yl)methyl acrylate)]; Tg: glass transition temperature.
To analyze the behavior of the relaxation times as a function of temperature in PACGA, data of DRA spectrum depending on the temperature at constant frequency were taken from the published literature. 23 The loss modulus at constant frequencies (1 Hz, 10 Hz, 102 Hz, 103 Hz, and 104 Hz) was analyzed to observe the glass transition shift since, in this work, the properties of kinetic rigidity were not calculated; however, they can be obtained with DRA data.
The graphic representation of this experimental behavior is shown in Figure 6 together with a “nonlinear” regression to obtain Vogel’s parameters. In this regression, the relaxation time at infinite temperature was considered as invariant with the value Bα = 5 × 10−12 s. The result obtained was D = 1.43 ± 0.001 and T0 = 260.85 ± 0.02 K, with a χ2 = 1.62 × 10−6. On the other hand, the Tg reported by DSC is equivalent to 281 K. 23
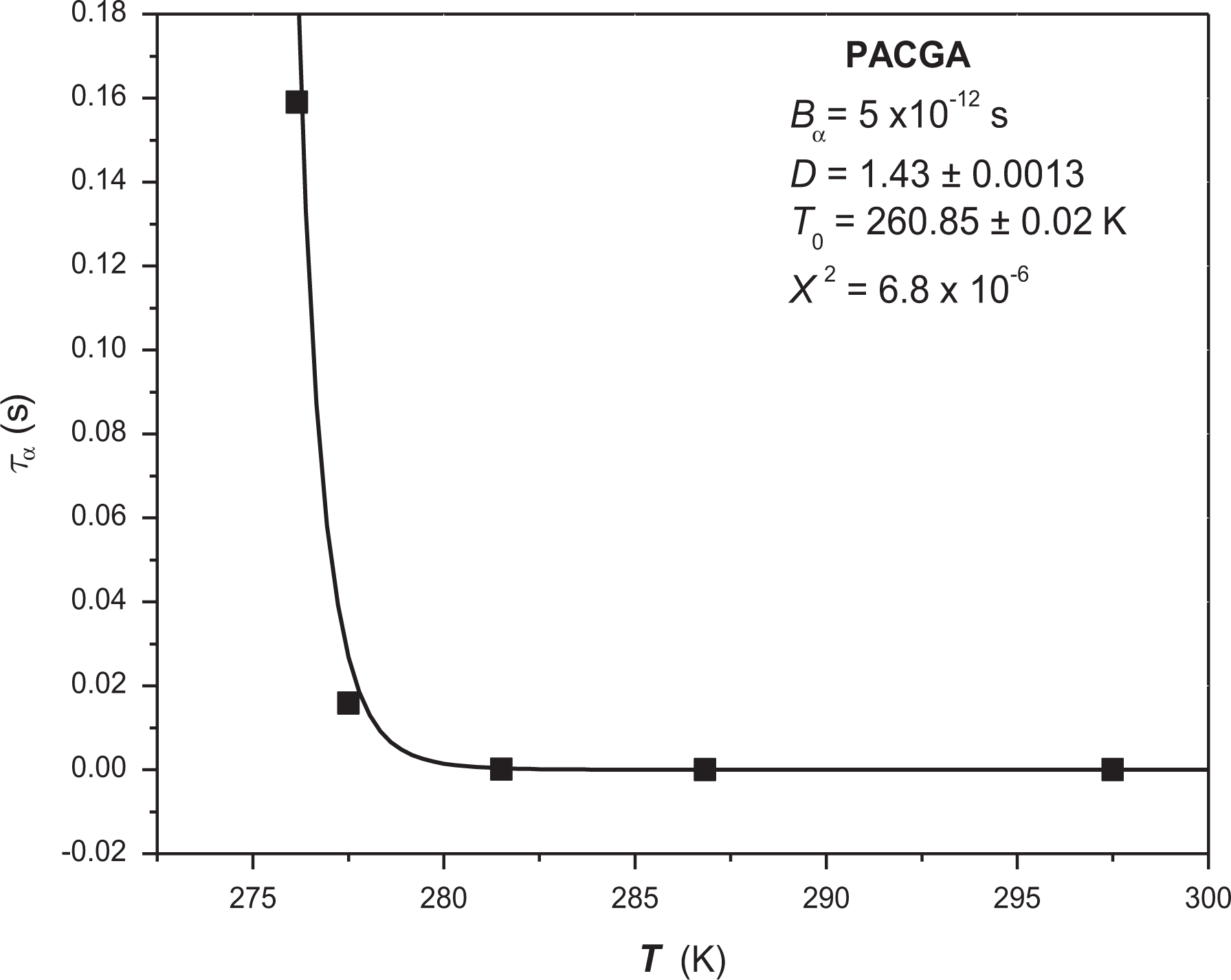
“Nonlinear” regression for α-relaxation of PACGA.
With this result, it is possible to observe that the structural modification of PMMA to obtain PACGA gives the result as a substantial increment in structural rigidity D that goes from 15.5 to 1.43, which demonstrates, as in other works, that substitutions with aliphatic groups can help to render the structure more flexible. Moreover, as the Vogel’s temperature obtained in the regression was T0 = 260.85 K, the use of this material in applications below T = –12°C is not recommended because structural mobility below such temperature is inhibited.
Diglycidyl ether of bisphenol-A
DEGEBA is the base of an epoxy resin and is also the result of research to optimize the mechanical properties of this kind of materials. 24 One of the factors for studying this material is its rigid behavior since generally, epoxy resins have high rigidity. During the last few years, the option has been to incorporate rubber in the chemical structure; however, with the use of rubber, the elastic modulus drops. According to other works about structural rigidity, the presence of benzene rings limits molecular flexibility. 16 DEGEBA presents a highly aromatic structure, as can be observed in Figure 7. The high aromaticity is one reason to expect that this material should present low levels of molecular flexibility, and therefore, it will have a low D parameter.
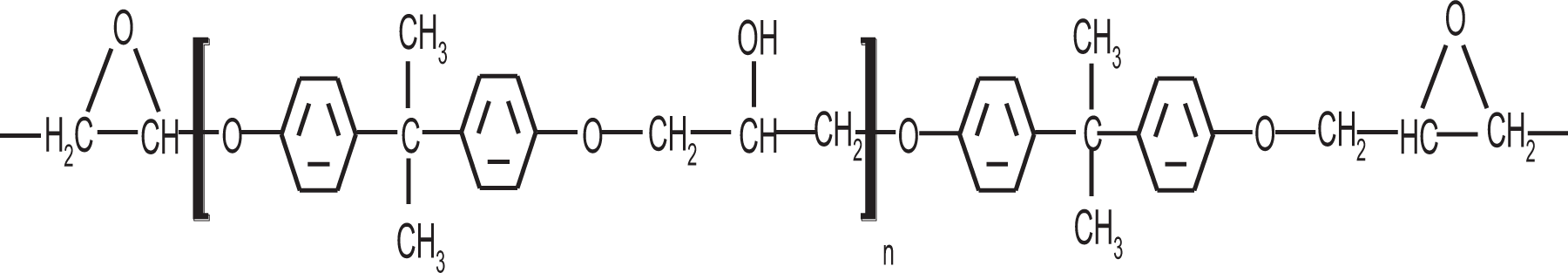
Chemical structure of DEGEBA.
A study that shows the variation in the loss modulus as a function of frequency at constant temperatures equivalent to 353, 343, 333, 323, 313, 303, 298, 293, 288, 283, and 278 K in the process of dielectric relaxation for DEGEBA has been already presented 24 ; however, data regarding the Vogel’s parameters were not included.
Taking the maximum for the main transition in the dielectric loss modulus ε″ as a function of frequency for DEGEBA at different temperatures, it is possible to determine the relaxation time for the glass transition τg that corresponds to each one of the evaluated temperatures, such data are reported in Table 4.
Experimental data of the α-relaxation of DEGEBA.
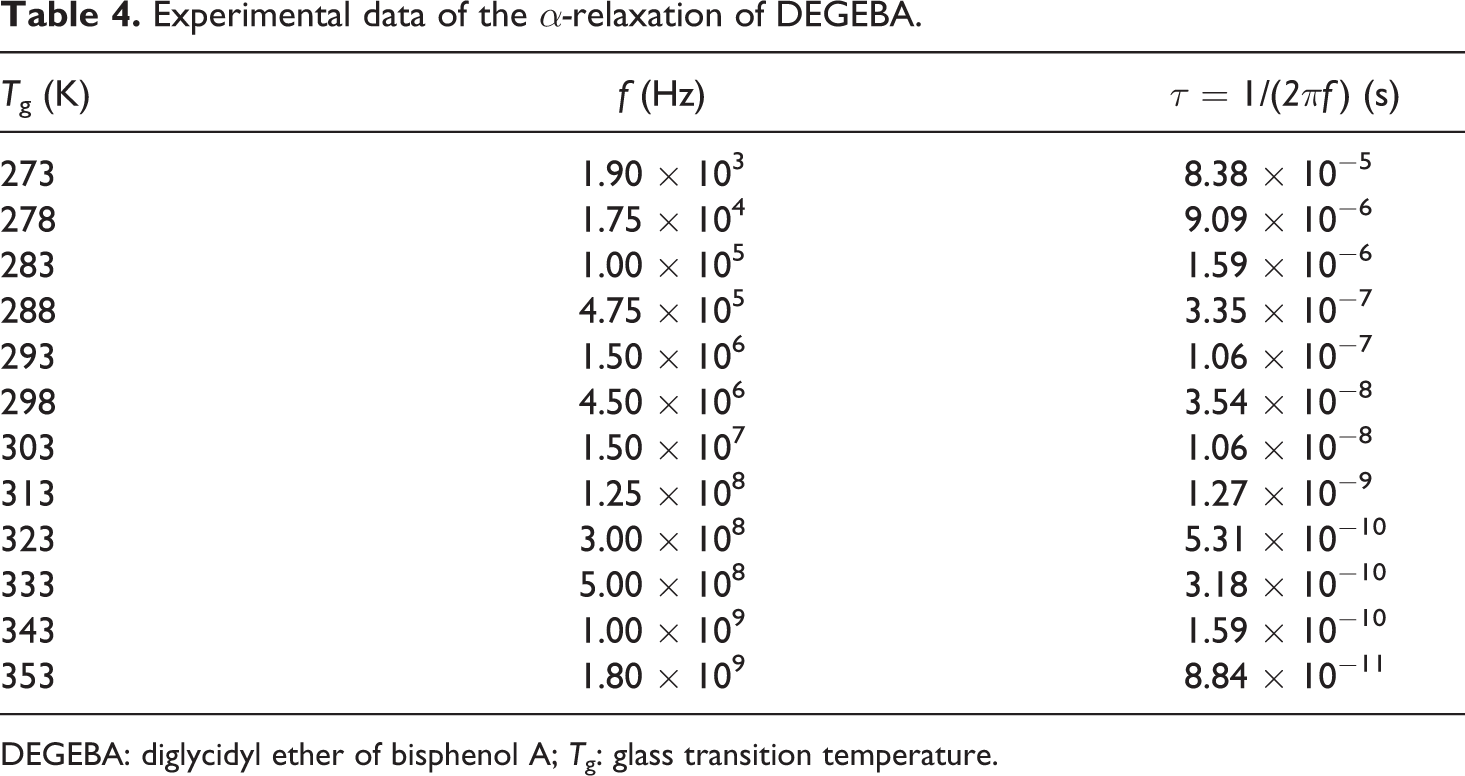
DEGEBA: diglycidyl ether of bisphenol A; Tg: glass transition temperature.
Analyzing these data, it can be observed that the glass transition shift agrees with the time–temperature superposition principle, stating that an increment in temperature has the same effect as if there were an increment in frequency or a decrease in the time over the measured viscoelastic behavior.
The “nonlinear” regression using Vogel’s model for DEGEBA gave as a result the following parameters: Bα = 5 × 10−12 s, D = 3.067, and T0 = 233.34 K. In this case, χ2 = 1.25 × 10–8. Figure 8 shows a continuous line that represents the regression curve with the parameters.
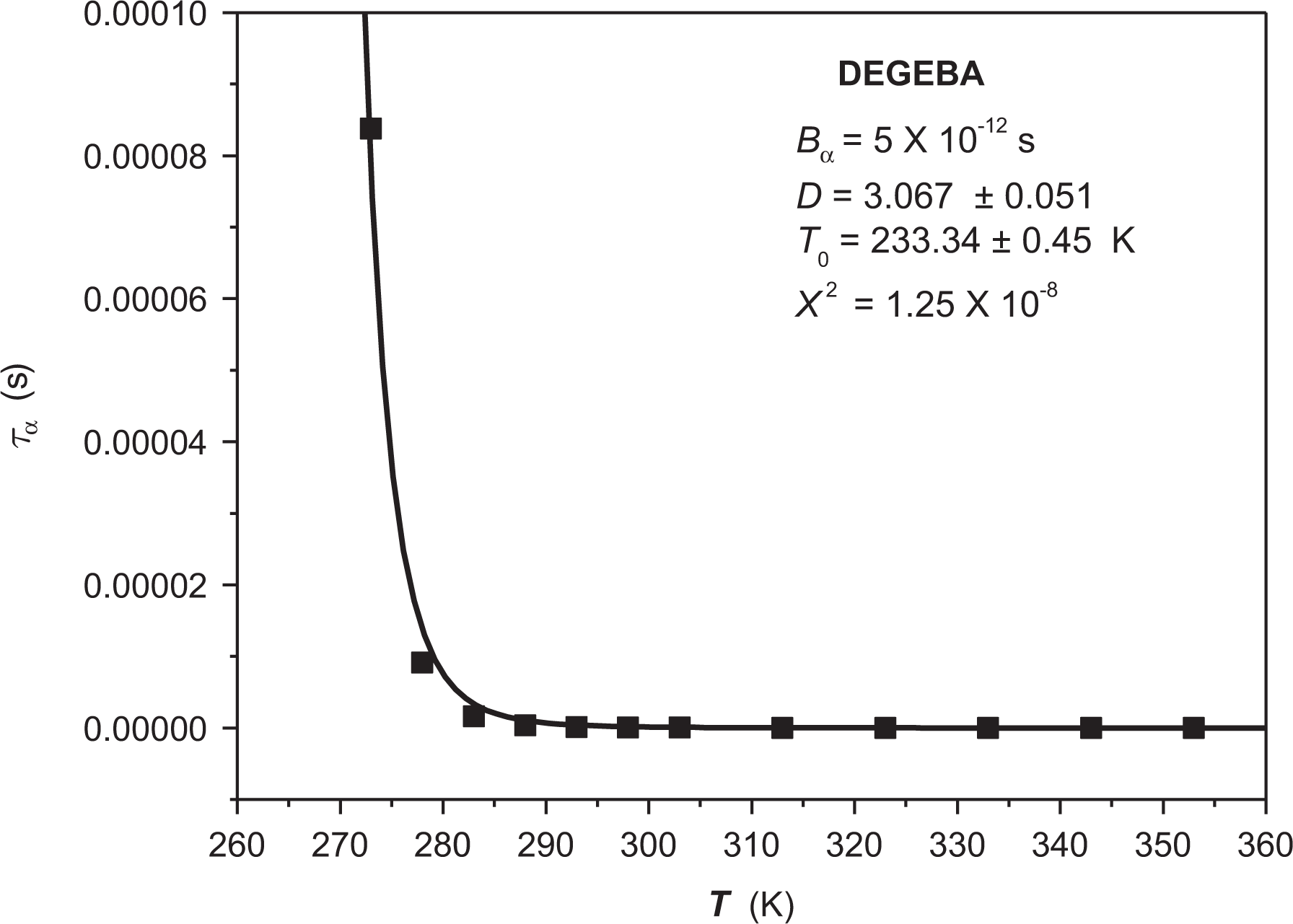
“Nonlinear” regression for α-relaxation of DEGEBA.
From these results, it is possible to conclude that DEGEBA is a highly rigid structure in comparison to those in Table 1. It is possible to say that this result matches what was stated before regarding the effect of aromatic groups over kinetic rigidity, with the phenyl groups responsible for the high level of rigidity. On the other hand, the obtained T0 value indicates that below −40°C, structural mobility in this material will be quite low, resulting in poor relaxation properties. Considering that Tg reported by DSC is equivalent to 257 K, 25 we can calculate the activation energy of the glass transition for this structure.
Activation energies for α-transition
A parameter that characterizes the behavior of polymer’s relaxation and that corresponds to the α-relaxation process is the constant C that results from the linear relationship of parameter D as a function of the dimensionless ratio Tg/T0 as expressed in equation (7), where Tg will represent the glass transition temperature reported through DSC.
Due to the fact that in this work we only reported specific data of three structures, it is necessary to complete such data with the other structures reported in Table 1 to limit the dispersion of the regression of D as a function of Tg/T0 and increase the data for the calculation of constant C. Figure 9 shows this linear regression where it is found that the constant C has a value 30.46.
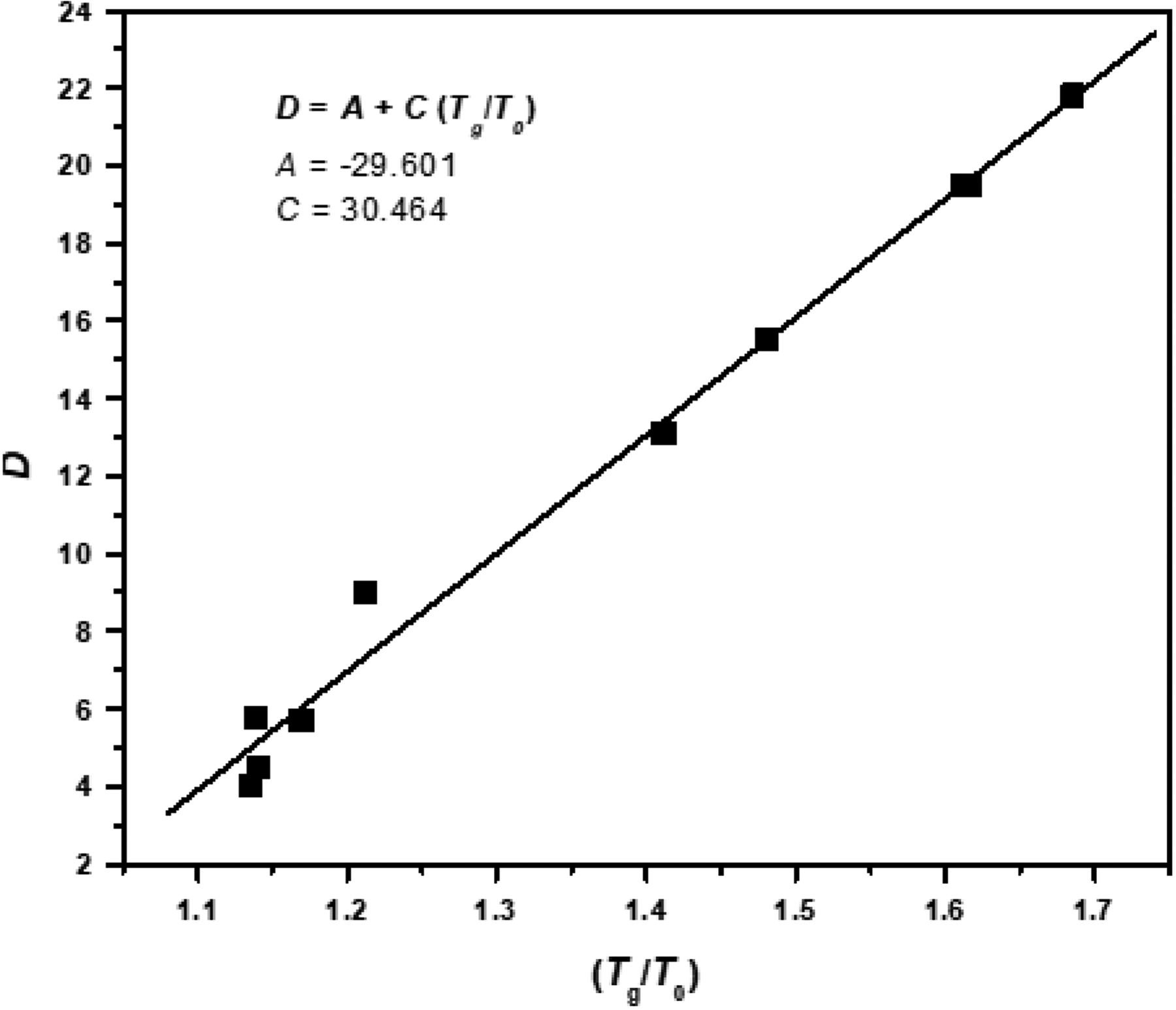
Linear adjustment of rigidity D as a function of the ratio Tg/T0.
Some studies show that the constant C for inorganic glassy materials is C = 36.8,
20
while as for flexible chain polymers (
Moreover, with Vogel’s parameters determined in this study, it is possible to obtain the value of the activation energy at infinite temperature, U∞. This energy is obtained when the temperature that corresponds to the maximum of α-transition is high (T → ∞), and it is related to the limiting value or the activation energy Uα(T), which characterizes its minimum value due to the fact that when T → ∞, the structure becomes more flexible, which means less energy is needed to carry out the relaxation process.
Equations (6) and (8) define both, the activation energy that corresponds to Tg determined by DSC (Uα(Tg)) and the activation energy U∞ respectively, with these relationships Uα (Tg) = CTgk and U∞ = DT0k. In these equations, the parameter k corresponds to the Boltzman’s constant.
According to Uα(Tg) results, the polyimide structure (β-CN) APB/ODPA is the material that requires the higher levels of energy to carry out the relaxation process in the glass transition. While DEGEBA is the one that requires a lower level of energy comparatively. Table 5 presents the data obtained for the activation energies as well as the parameters used to determine them.
Activation energies of structures.
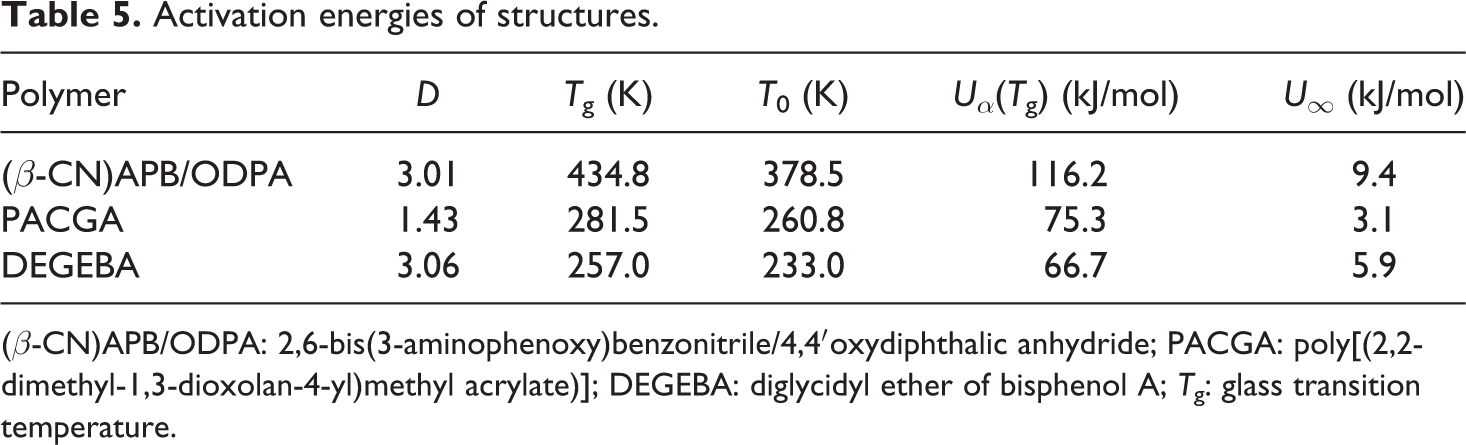
(β-CN)APB/ODPA: 2,6-bis(3-aminophenoxy)benzonitrile/4,4′oxydiphthalic anhydride; PACGA: poly[(2,2-dimethyl-1,3-dioxolan-4-yl)methyl acrylate)]; DEGEBA: diglycidyl ether of bisphenol A; Tg: glass transition temperature.
The value of Uα(Tg) for the structure (β-CN) APB/ODPA is equivalent to 116.31 kJ/mol; for the PACGA structure, it was 75.26 kJ/mol; and for DEGEBA, was 66.68 kJ/mol; while the value corresponding to U∞ for the structure (β-CN) APB/ODPA was 9.44 kJ/mol, for PACGA it was 3.09 kJ/mol, and for DEGEBA was 5.92 kJ/mol.
It is also found that PACGA presents the lowest level of energy to carry out the α-transition and also has more flexible linkages. It should be considered that even though the structural mobility is detected by DRA, Vogel’s model gives information about the interval of temperature in which there exist molecular motions for relaxation independently of the magnitude of these movements.
This model can help to obtain information on the interval of temperatures at which amorphous rigid materials can be useful, this is because below the interval T0 < T < Tg, the mechanical properties of the polymer are poor due to the lack of ability to perform molecular relaxations. The information regarding the magnitude of chain mobility in relation to mechanical properties at a specific temperature is contained in the storage modulus E′.
Finally, the behavior of activation energy U∞ as a function of the interval of structural mobility (Tg–T0) is presented in Figure 10. This figure shows the behavior of the activation energy U∞ as a function of the interval of structural flexibility for the materials that were studied in this work, which clearly indicates a correlation of larger values of U∞ as the interval of temperatures increases.
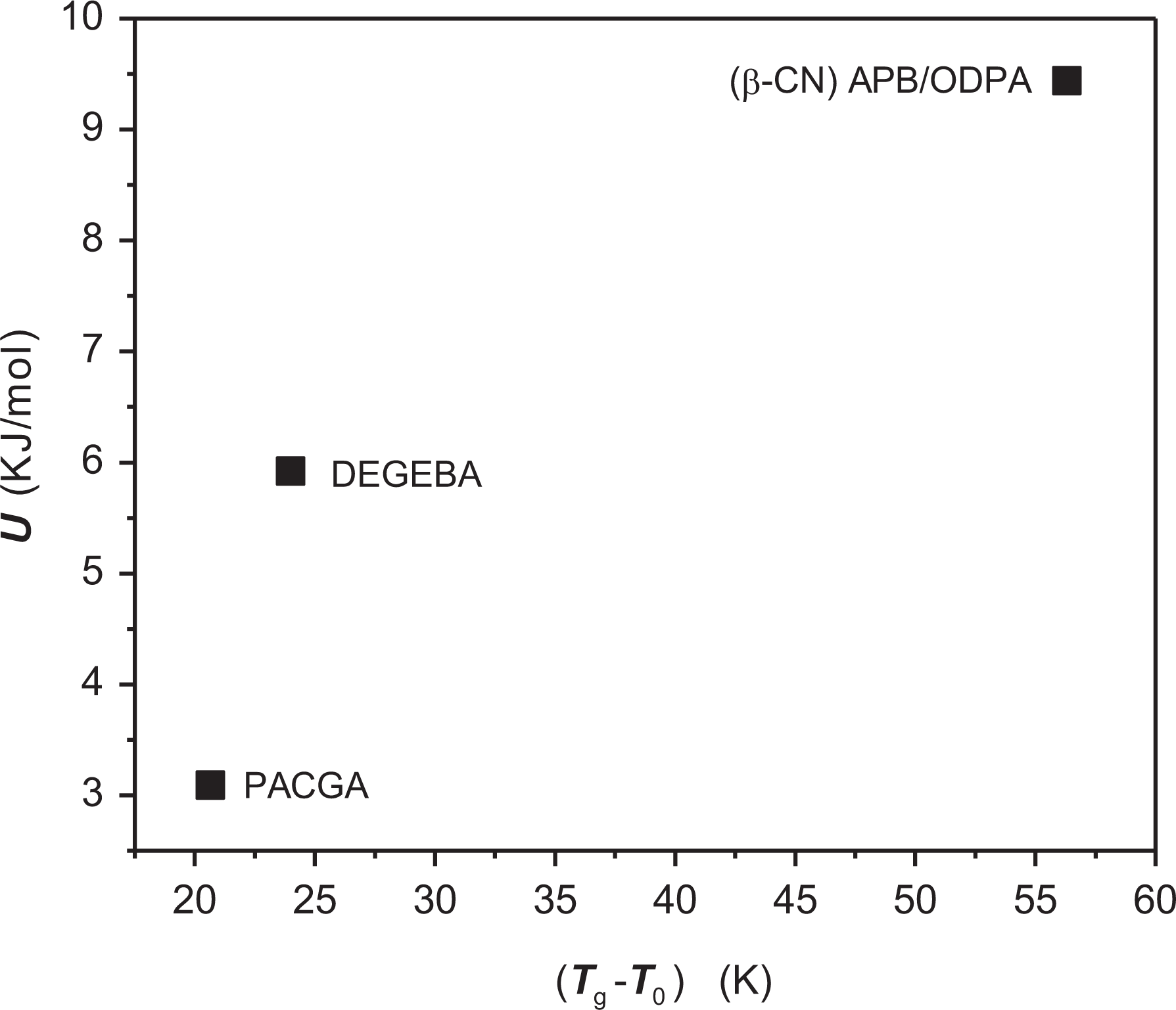
U∞ as a function of the interval of temperatures of structural mobility (Tg – T0) for the polymers of this work.
To generalize the results, Figure 11 shows the behavior obtained for the polymers reported in Table 1. Based on both graphics, it is possible to establish that as the interval of structural mobility increases, higher levels of energy will be required to carry out the relaxation process at the glass transition.
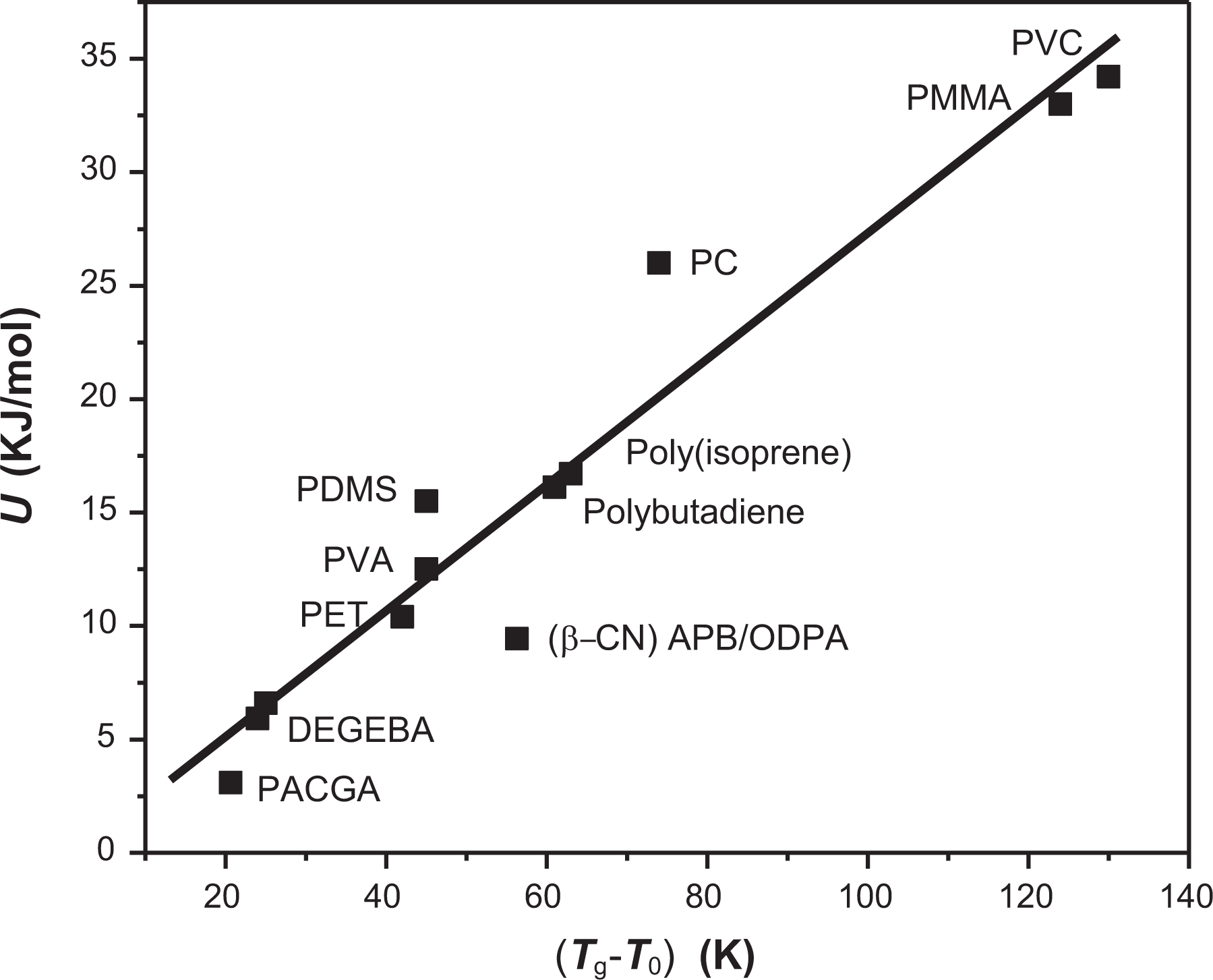
U∞ as a function of the interval of temperatures of structural mobility (Tg – T0) for polymers in Table 1.
Conclusions
The analysis of the kinetic rigidity on the three structurally different materials confirms the importance of analyzing the effect of chain structure on rigidity properties of polymers since it can be used as a tool for the design of new materials. In this study, it was found, due to a higher energy to effect the glass–rubber transition, as other researchers have pointed out, that the aromatic rings impart high levels of rigidity to the structure, as found for (β-CN) APB/ODPA and DEGEBA, while the aliphatic functional groups confer flexibility, as in the case of PMMA and PACGA which was analyzed in this work.
On the other hand, the linear regression for the relationship between the rigidity parameter D as a function of the ratio Tg/T0 found that constant C of the model is equivalent to C = 32.4. This result is not in agreement with the ones published in other works, where it is stated that this constant is C = 39 for flexible polymers (
Moreover, when Vogel’s temperature value is used, it is possible to establish some working temperature range for the polymers in terms of their expected relaxation behavior. It can be stated, based on T0, that polyimide (β-CN) APB/ODPA is not recommended for applications below 161°C. While the acrylic PACGA is not recommended for applications that require high chain mobility below 8°C, and bisphenol-A DEGEBA is not recommended for such applications below −16°C.
These recommendations came from the fact that Vogel’s temperature T0 limits the use of polymeric systems since below this temperature the structural mobility properties are inhibited and then relaxation properties are poor.