
Abstract
Degradable poly(ester urethane)s (PEUS)/nanosilica composites are prepared, and a preliminary evaluation of their potential to be used in calcified tissue regeneration is performed. First, poly(ethylene glycol succinate) (PEGS) of different molecular weights is prepared and then a prepolymer with an excess of 1,6-hexamethylene diisocyanate is synthesized; this prepolymer is subsequently extended with 1,4-butanediol in the presence of nanosilica particles. The effects of the structures of PEGS and PEUS are studied by means of attenuated total reflectance infrared, gel permeation chromatography, X-ray diffraction, thermogravimetric analysis, optical microscopy, and scanning electron microscopy. The materials show that similar crystalline structure independently of the molecular weight, however, increases the thermal resistance with higher molecular weight of nanocomposites. After soaking in simulated body fluid, the appearance of apatite phosphate bands in Fourier transformed infrared spectra suggests the bioactive character of these composites. In addition, degradation and toxicity test are performed. The materials are degradable but not cytotoxic after 7 days of testing.
Introduction
Biodegradable polymers such as aliphatic polyesters and their copolymers have drawn growing attention from both academic researchers and industry due to their potential applications in the biomedical materials field and as environmentally friendly materials. 1 An economical and effective way to improve the required properties of polyesters for many applications is throughout reaction with other reactives. A typical example of this method is the synthesis of poly(ester urethanes) (PEUS). PEUS can be tailored to meet the highly diversified demands of different modern technologies such as coatings, fibers, foams, and thermoplastic elastomers. 2 The properties of these PEUS are strongly dependent on the molar mass and the polymolecularity of the soft segment component 3 as well as on the chemical structure of all the components. 2 Several PEUS based on segments of polylactic acid, 4 –6 polycaprolactone, 7,8 polyethylene oxide, 9,10 or copolymers of them 11 have been manufactured for biomedical applications.
Several attempts have been made to spread PEUS application into the field of substitution or regeneration of musculoskeletal tissue substitution or regeneration. 12 –14 Other studies tailored the nanofiber size of the polymeric matrices to be in the range of 180–650 nm, to mimic the architecture of the extracellular matrices (ECMs) 13 or supplement the PEUS with mineralized ECM laid by human mesenchymal stromal cells to enhance the biological performance of the materials, 14 but very few attempts have been made to induce bioactivity to a PEU-based system intended for musculoskeletal tissue regeneration. 12
Bioactivity is the ability of certain materials to promote a direct bonding with the hosting tissue. 15 Several studies have shown that bioactivity is critical for the integration between a living tissue and a biomaterial. 16,17 Some ceramics and glass ceramics such as hydroxyapatite, wollastonite, or Bioglass® have shown a good ability to bond to living bone through the formation of biologically active bone-like apatite. However, the mechanical properties are one of the factors that prevent them from not extensive use as bone substituted or as soft tissue replacement. 18 –20
By other way, organic–inorganic hybrid materials have been used as support for bond the living bone with the material, because the organic character (polymer) gives them a good mechanical resistant, and the inorganic (ceramic or glass ceramic) gives the bioactive character to this materials. The problem in this kind of materials is its raw materials that are in some cases expensive and their synthesis is complicated. 21
Using composite materials with a polymeric matrix and bioactive ceramic or glass ceramic could be an alternative to have bioactive materials with mechanical properties enhanced. The production of nanocomposites with nanoparticles dispersed in a polymer matrix has the potential to improve interaction with host tissue/cells. 22,23 The main problem with composite materials is the polymers used. Conventional polyesters degrade very quickly once hydrolysis begins or there may be polymers with suitable rate of degradation but their hydrolysis can also yield toxic by-products. 24
The aim of this work is to prepare PEUS/nanosilica particle composites and to perform a preliminary evaluation of their bioactivity and biotoxicity in order to evaluate the potential to be used for calcified tissue regeneration without yielding toxic by-products.
Experimental section
Reactants
Poly(ethylene glycol) Mw = 1500 and 3500 g mol−1 (PEG1500 and PEG3500; Sigma-Aldrich, Missouri, USA) and succinic acid (SA; 99%+ purity, Sigma-Aldrich) were used to prepare the polyesters PEGS1500 and PEGS3500. 1,6-Hexamethylene diisocyanate (HDI; 98% purity, Sigma-Aldrich), 1,4-butanodiol (BD; 99% purity, Sigma-Aldrich), and Aerosil® OX 50 (silicon dioxide (SiO2), average primary particle size of 40 nm, specific area 50 m2 g−1; Degussa, Germany) were used together with the polyesters to synthesize polyurethanes. Tin (II) 2-ethylhexanoate (OctSn; approximately 95% purity, Sigma-Aldrich) was used as a catalyst, and N,N′-dimethylformamide (DMF; extra pure, Sigma-Aldrich) was the solvent used for the synthesis of the polyurethanes.
Synthesis of poly(ethylene glycol succinate)
Poly(ethylene glycol succinate) (PEGS) was prepared by the melt polycondensation method using an SA–PEG molar ratio of 1.2:1.0. The procedure is based on that described in the studies by Nivasu et al. 25,26 ; 20 g of PEG (0.013 mol) and 1.9 g of SA (0.016 mol) were added to a three-necked round-bottom flask equipped with a nitrogen inlet, a distillation condenser, and a magnetic stirrer. The temperature was slowly raised to 180°C and maintained for 4 h. The water formed during the reaction was collected in a round-bottom flask.
Synthesis of PEU/silica nanocomposites
The PEUS were synthesized in a two-step process. In the first one, 5-wt% SiO2 nanoparticles were dissolved in DMF by sonication with a total energy of 6 kW·s for 12 min. This solution was added to 30 mL of a DMF solution containing 5.04 g of PEGS (2 mmol) and 0.1 wt% of OctSn. The mixture was performed at 100°C. After cooling up to 60°C, 0.66 mL of HDI (4 mmol) solved in 5 mL of DMF was added and the resulting mixture heated at 86°C for 2 h under a nitrogen flow. In the second step, 0.177 mL (2 mmol) of the chain extender (BD) solved in 5 mL of DMF was added into the prepolymer and then the reaction proceeded for 2 h. The DMF was eliminated under reduced pressure at 90°C for 24 h.
PEG molecular weight, polyesters codes, mole ratio of reactants are shown in Table 1, and PEUS molecular weight, molar taio, and codes are shown in Tables 2.
PEG molecular weight (Mw ) and PEG:SA molar ratio used in the synthesis of the polyesters.a
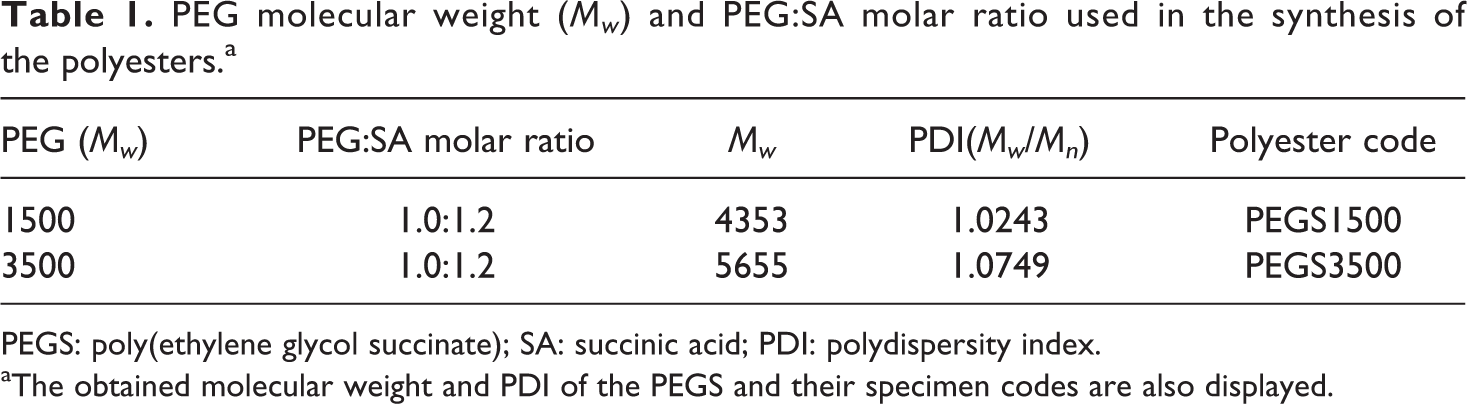
PEGS: poly(ethylene glycol succinate); SA: succinic acid; PDI: polydispersity index.
aThe obtained molecular weight and PDI of the PEGS and their specimen codes are also displayed.
PEGS selected for the synthesis of PEUS, HDI–PEGS–BD ratio, and percentage of SiO2 used in the reactions.a

PEGS: poly(ethylene glycol succinate); PEU: poly(ester urethane); HDI: 1,6-hexamethylene diisocyanate; BD: 1,4-butanodiol; PDI: polydispersity index; SiO2: silicon dioxide.
aMolecular weight and PDI obtained for the PEU and their respective specimen codes are also displayed.
Materials characterization
Proton nuclear magnetic resonance (1H-NMR) spectroscopy was performed on a Varian Inova 300 spectrometer (Palo Alto California,USA) operating at 300 MHz and at room temperature, using deuterated chloroform as a solvent.
Attenuated total reflectance-Fourier transformed infrared (ATR-FTIR) spectra were registered using a PerkinElmer Spectrum One (Connecticut, USA) in the range of 4000–650 cm−1, with a 16 scan accumulation and using a 2 cm−1 resolution.
Wide-angle X-ray diffraction (WAXD) patterns were obtained using a Brucker X-ray diffractometer (Karlsruhe, Germany) operating at a rate of 2° min−1 from 10° to 60° in 2θ. Thermogravimetric analysis (TGA) diagrams were obtained in a TGA-Q500 (TA Instrument, New Castle, USA) apparatus in a range of 30–800°C at a heating rate of 5°C min−1 under a nitrogen atmosphere.
The average molecular weight (Mw ) and polydispersity index (Mn /Mw ) of polymers were determined by gel permeation chromatography using a Waters 244 gel permeation chromatograph equipped with a refractive index detector. Polystyrene standards were used for calibration and Tetrahydrofuran, HPLC-grade was used as mobile phase at a 1 mL min−1 flow rate.
Degradation was tested by soaking specimens (10 × 10 × 5 mm) into 30 mL of phosphate buffer solution (PBS) and the specimens were incubated at 37°C. The weight loss and pH were measured after 1, 2, 4, 8, 15, and 28 days. Bioactivity in vitro test of the composite was followed by the growing of a hydroxyapatite layer on the surface of soaked material in simulated body fluid (SBF).
The cytotoxicity was analyzed with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, 27 using human osteoblasts from the European collection of cell culture. Cell growth was performed in a controlled atmosphere using Dulbecco’s modified Eagle’s medium modified with 4-(2-hydroxyethyl)-1-piperizine ethanesulfonic acid and supplemented with 10% fetal bovine serum and penicillin-streptomycin antibiotic solution. The toxicity of extracts eluted from PEUS composites was determined as follows. Thermanox® (TMX) control disc and PEUS material discs were placed in 5 mL of modified eagle’s medium (MEM) and incubated for 24, 48, and 72 h. After the end of contact, the PBS was replaced with 100 µL/well of MTT-sodium succinate in PBS and they were incubated at 37°C for 4 h. During this time, mitochondrial dehydrogenase, present in live cells, transformed the yellow MTT solution into insoluble blue formazan. Thus, cell viability was estimated by measuring the amount of formazan produced. At the end of incubation period, the excess of MTT was removed and 100 µL of dimethyl sulfoxide was added to dissolve the formazan. The plates were swirled for 20 s to high intensity until the purple color becomes uniform and the absorbance was evaluated at 570 nm. Analysis of variance of the results was performed with respect to TMX at a significant level α < 0.05.
Results and discussion
PEGS was synthesized following the reaction scheme displayed in Figure 1(a). The appearance of PEGS1500 and PEGS3500 obtained is white solids. The 1H-NMR for PEGS1500 is shown in Figure 2(a). The spectrum for PEGS3500 is similar and it has been removed for clarity of the figure. The proton resonance signals from polyesters obtained are assigned as follows: the peak appearing at δ = 4.2 ppm was assigned to the methylene protons in the ester bonds (–COOCH2–), δ = 3.6 ppm was assigned to the methylene protons in the repeating units of PEG (–CH2–CH2–), and δ = 2.55 ppm was assigned to the methylene from the SA (–CH2CH2–).
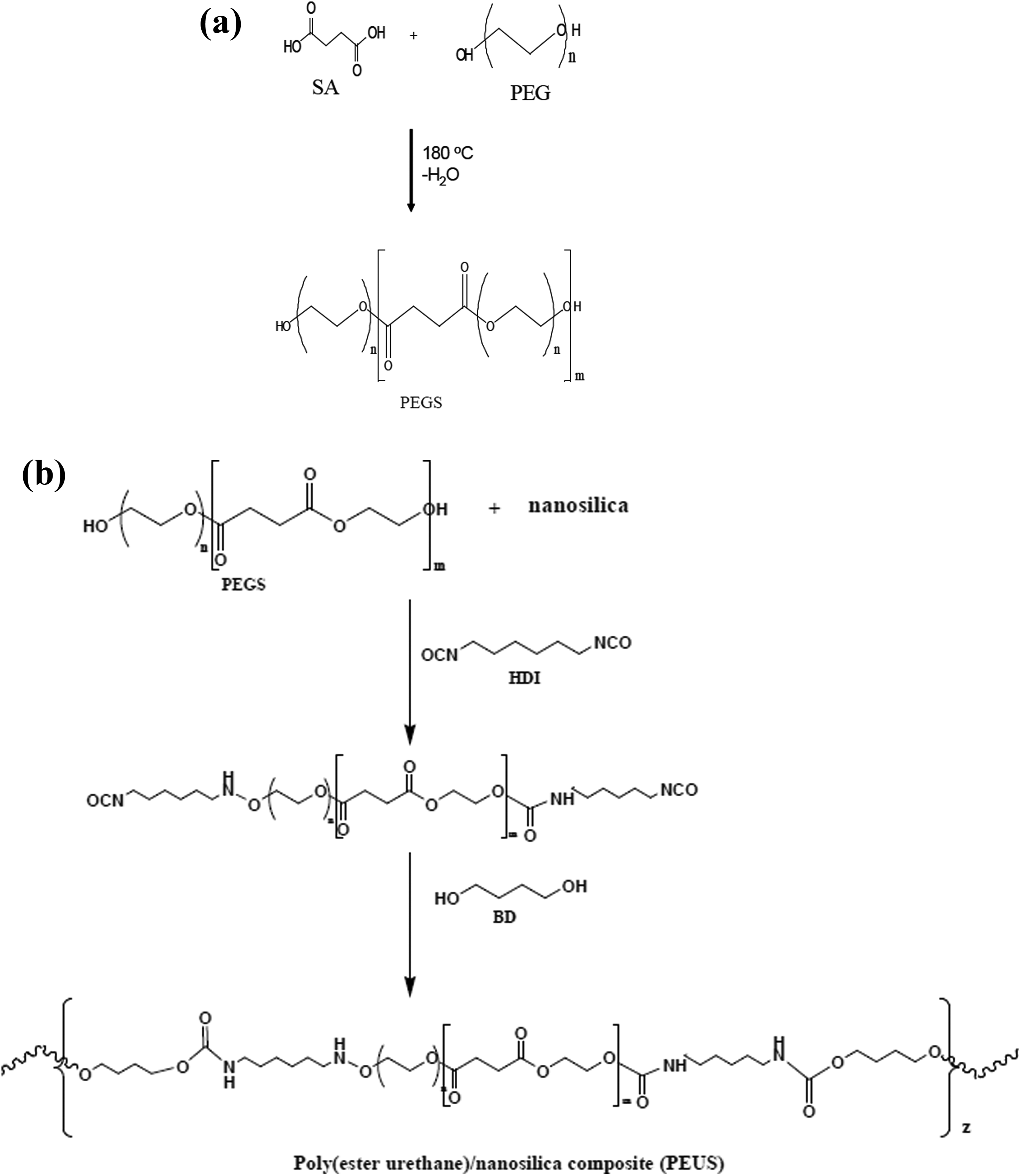
(a) Schematic representation of synthetic route for polyesters. (b) Schematic representation of synthetic route for polyurethane/SiO2 composites. SiO2: silicon dioxide.
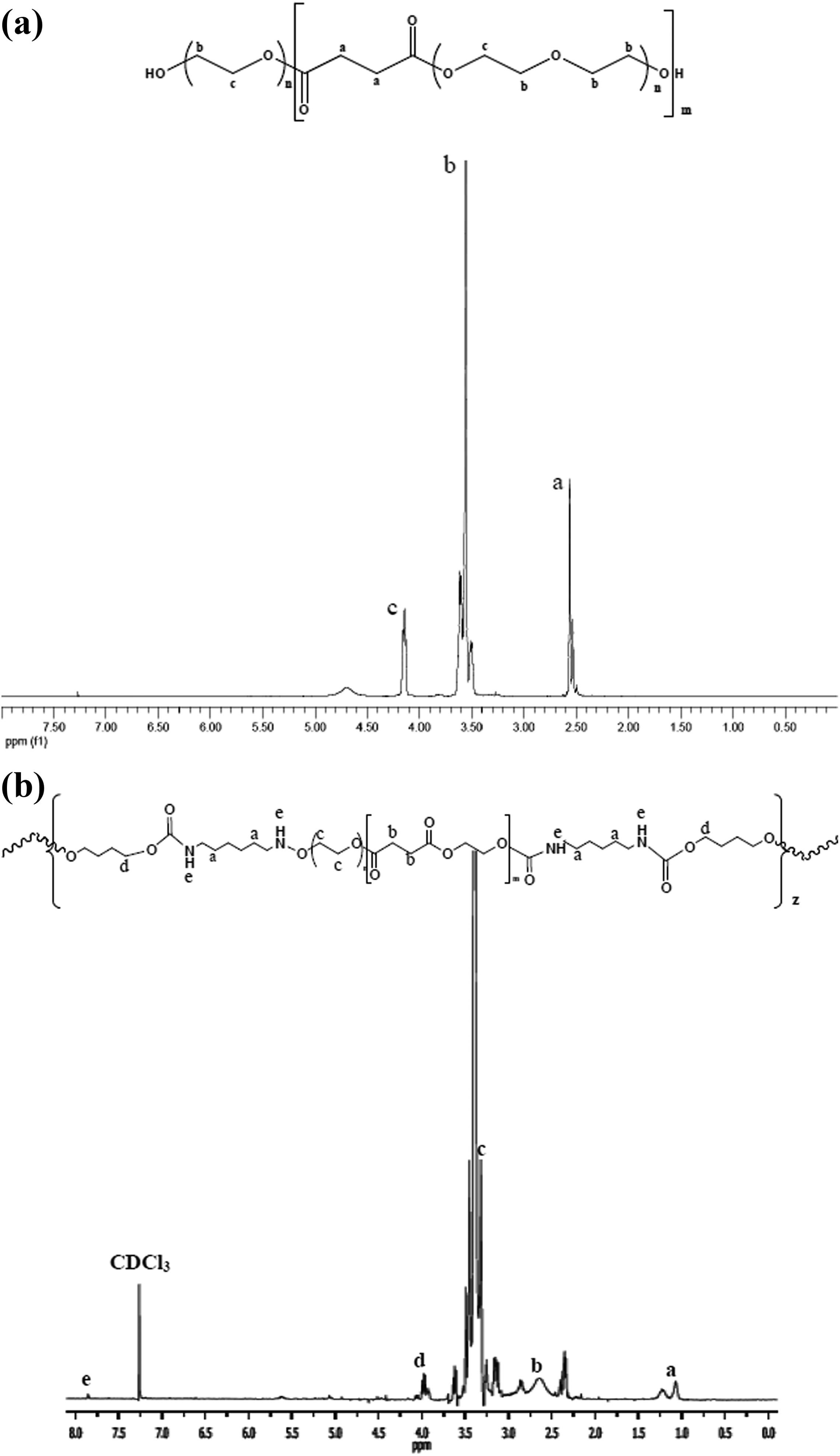
(a) 1H-NMR spectra of PEGS1500. (b) 1H-NMR spectra of PEUS1500. 1H-NMR: proton nuclear magnetic resonance; PEGS: poly(ethylene glycol succinate).
The ATR-FTIR spectrum of PEGS1500 is presented in Figure 3(a). The most significant bands appreciated from polyesters obtained are due to the carboxylic bonds at 1740 cm−1, the –C–O–C– stretching bands at 1100 and 1060 cm−1, and the –CH2– stretching band at 2880 cm−1.
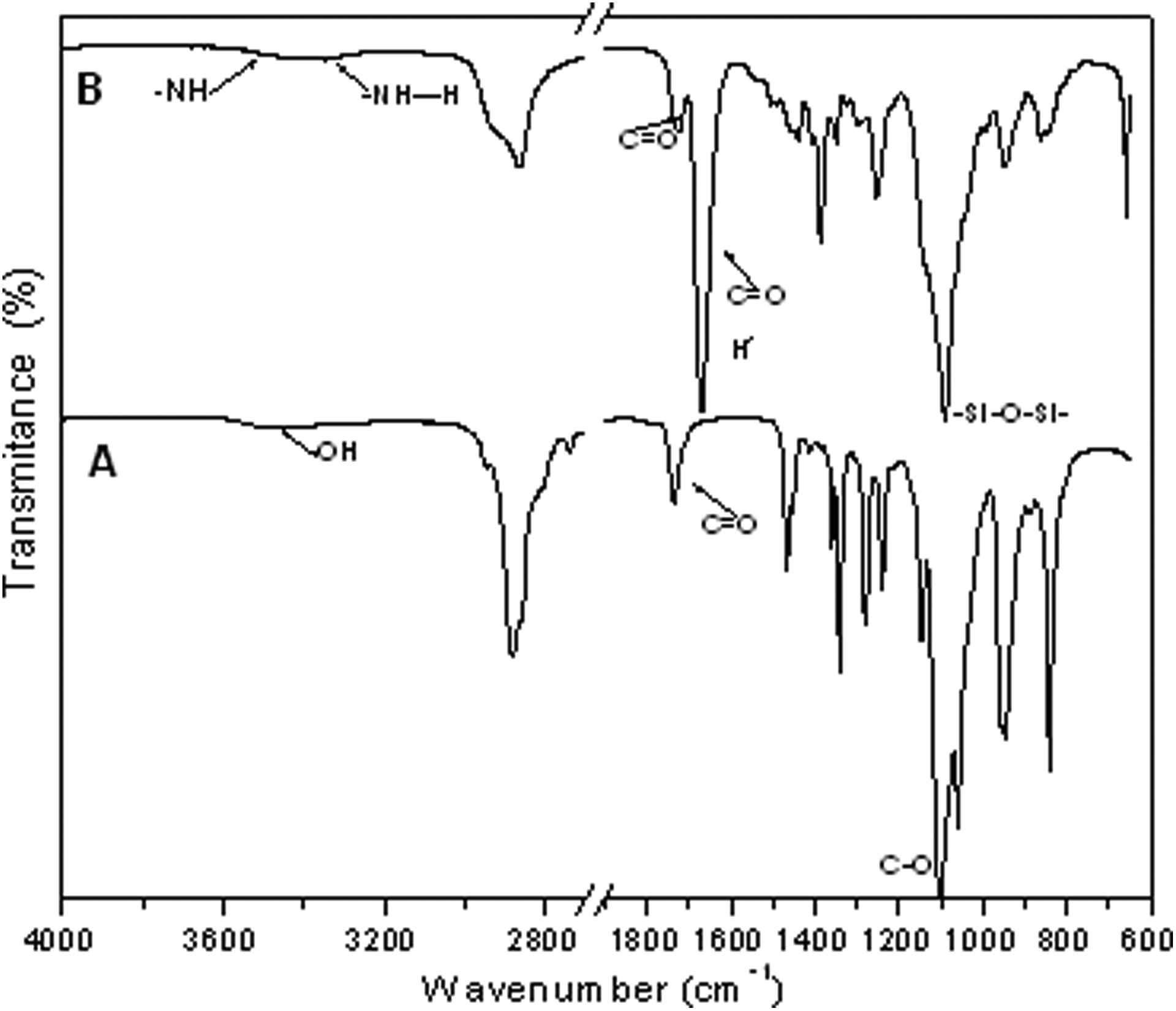
ATR-FTIR spectrum of (a) polyester and (b) composite. ATR-FTIR: attenuated total reflectance-Fourier transformed infrared.
The PEUS nanocomposites were synthesized in a two-step process by a reaction of the obtained PEGS with HDI and BD in the presence of a SiO2 solution as described in the experimental section. In Figure 1(b), the reaction scheme is displayed.
The 1H-NMR spectrum for PEUS1500 is shown in Figure 2(b). Peaks that were assigned to the PEGS component in Figure 2(a) also can be assigned for PEUS plus the peaks at δ = 1.0 ppm, δ = 1.25 ppm, and δ = 2.4 ppm that are assigned to the methylene from the isocyanate (–CH2CH2–). Also the contribution of proton bonding to nitrogen at δ = 7.8 ppm (–N–H) can be appreciated.
In Figure 3(b), the ATR-FTIR spectrum of the PEUS1500 is presented. The main differences from the PEGS spectrum displayed in Figure 2(a) are the bands at approximately 3451 cm−1 and at approximately 3340 cm−1 assigned to the free –N–H bonds and hydrogen bonded –N–H stretching, respectively. 28 The band at 1726 cm−1 is assigned to free –C=O and at 1671 cm−1 is due to the hydrogen bonded in –C=O groups for the PEU. 29 The hydrogen bonded due to a link between –NH and –C=O of the polyurethane and the interaction between nanosilica particles with the PEU 30 is located at 1630 cm−1. Additionally, it is observed the characteristic band at 1088 cm−1 attributed to the Si–O–Si bond and at 950 cm−1 ascribed to the Si–OH group of the nanosilica unreacted with the polyester. 21,31
WAXD patterns of the PEUS nanocomposites showed two broad diffraction peaks at 20° and 23° in 2θ The patterns of PEUS1500 and PEUS3500 are displayed in Figure 4. These materials showed a semicrystalline nature that it could be attributed to the semicrystalline nature of the polyethylene glycol. 32 A greater Mn of the PEG used in the reaction yields a greater crystalline character of the resulting PEUS nanoparticles reflected in a greater intensity of the mentioned diffraction peaks. However, it could be that the addition of nanosilica particles induces a lower crystallinity probably due to an increase in the degree of phase separation. 33
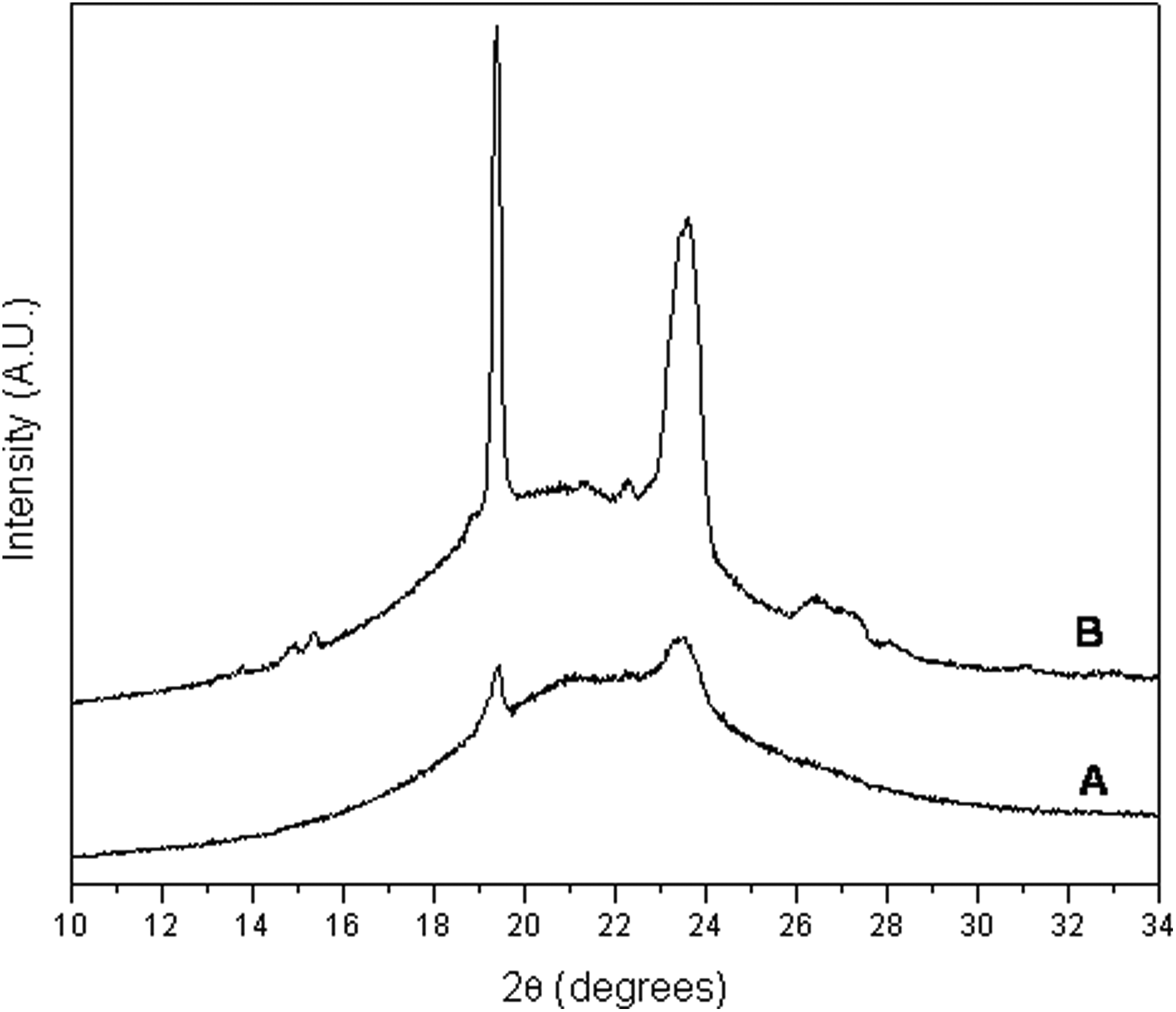
WAXD patterns of (a) PEUS1500 and (b) PEUS3500. WAXD: wide-angle X-ray diffraction.
The morphology of the PEUS nanocomposites was verified with optical microscopy as shown in Figure 5. A lamellar morphology can be observed with the appearance of platelets in the case of PEUS1500 that are not appreciated for PEUS3500. In order to see the dispersion of nanoparticles, a scanning electron microscopy (SEM) was performed, only the micrography of PEUS3500 is showed as example (Figure 6)a good dispersion and distribution of the silica nanoparticles it is observed.
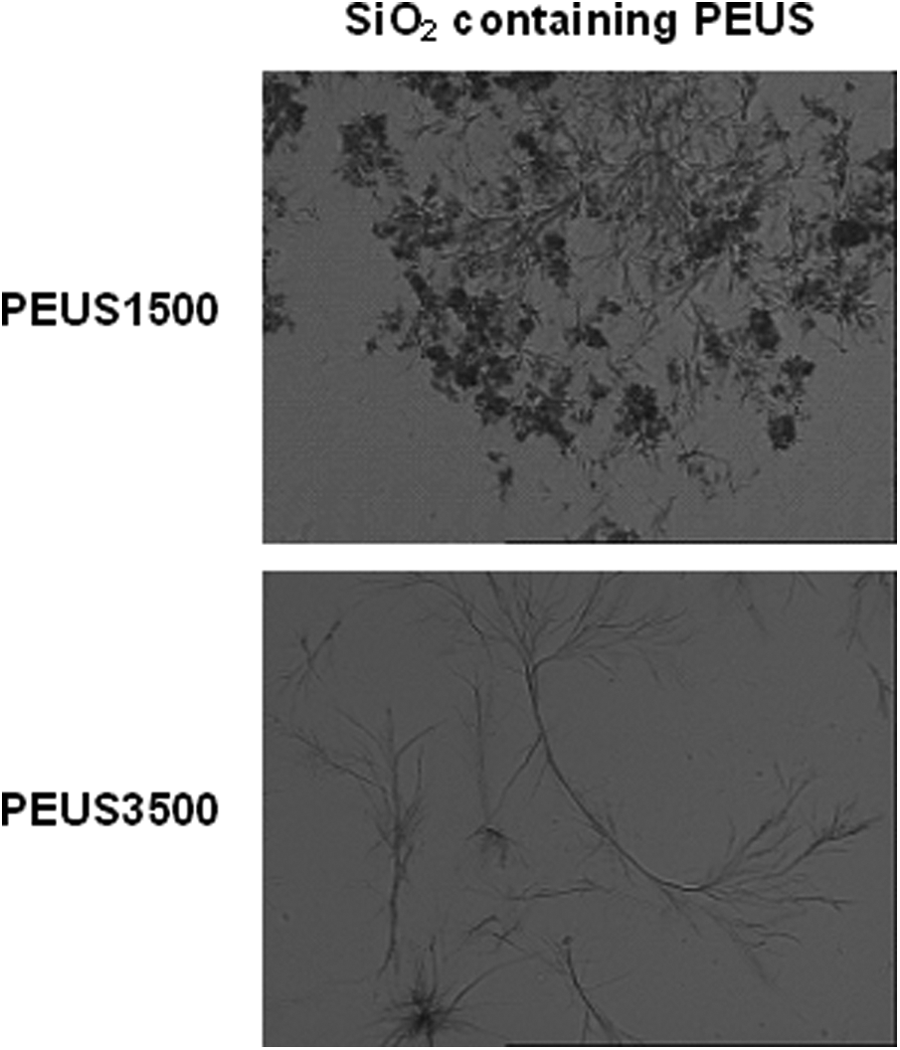
Optic microscopy image of PEUS1500 and PEUS3500.

SEM of PEUS3500. SEM: scanning electron microscopy.
The TGA of PEUS1500 and PEUS3500 is shown in Figure 7, and the weight loss below 200°C could be attributed to the evaporation of water physically absorbed onto the surface of material and can also be due to the allophanates and biuret that could have formed during the synthesis of the PUBs. 34 All the PEUS were degraded thermally in two steps. The first one can be attributed to the hard segment and yields alcohols and isocyanates. It occurs at approximately 342°C for PEUS1500 and at approximately 325°C for PEUS3500. The second decomposition process is due to the decomposition of the soft segment and it occurs at approximately 408°C for PEUS1500 and at approximately 412°C for PEUS3500. As it can be seen, the decomposition temperature is higher for higher molecular weight of the polymer. The total weight loss was 95% and it can be related to the amount of silica added (5%).
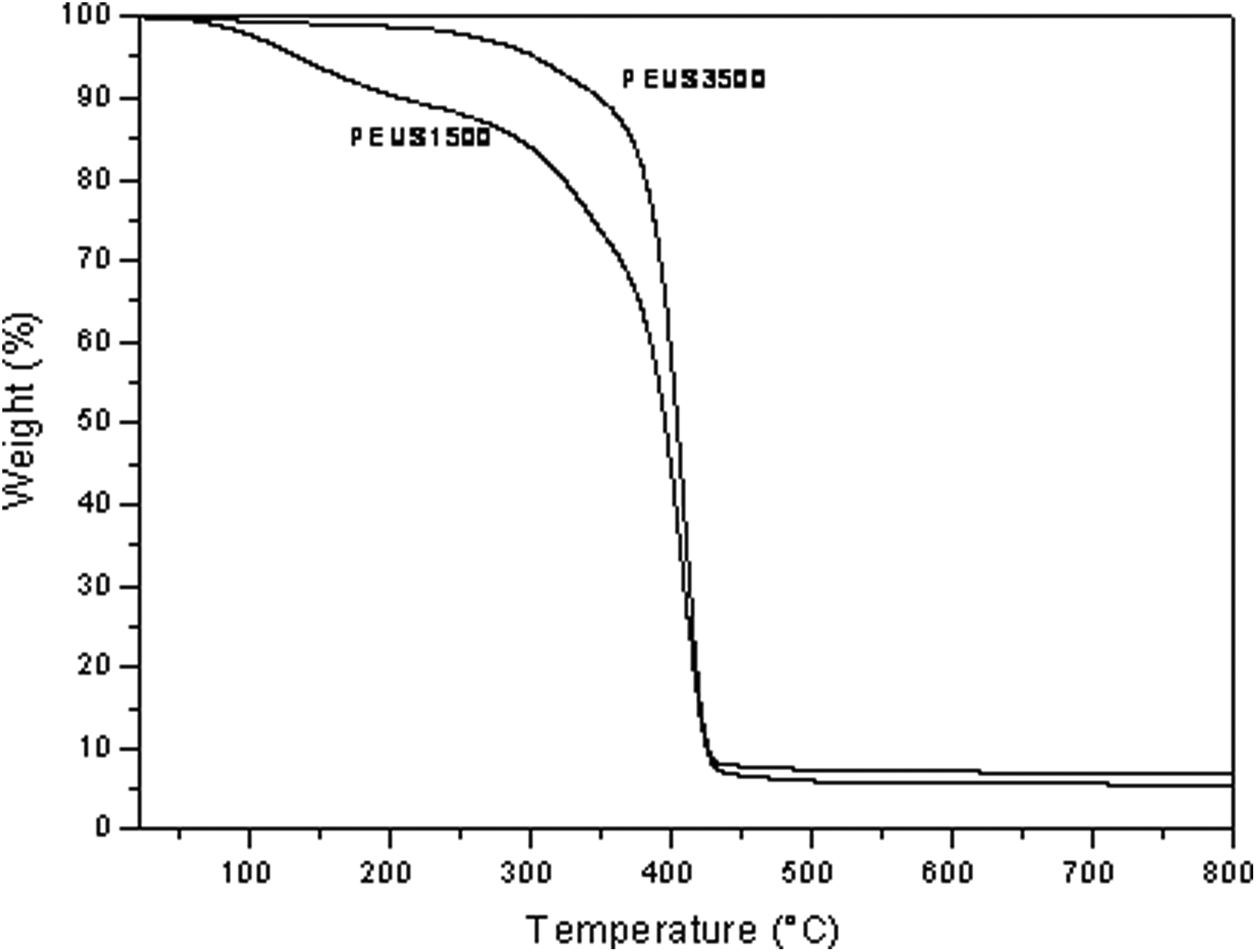
TGA curves of PEUS1500 and PEUS3500. TGA: thermogravimetric analysis.
The pH change on PBS was induced when PEUS1500 and PEUS3500 specimens are soaked on this buffer solution; the pH behavior of the solution is over several days as shown in Figure 8. It can see that the behavior was the same for both materials (PEUS1500 and PEUS3500). During the first 5 days, the pH of PBS decreases. This decrease could be attributed to the release of acidic groups from the polyester segment. 35 After 8 days, the pH begins to increase perhaps due to the start of the degradation of the hard segment, forming amines and carbon dioxide.

pH change of PBS solution at different times for PEUS1500 and PEUS3500. PBS: phosphate buffer solution.
The PEUS material behavior soaked in SBF can be follow by infrared spectroscopy with the disappearance of the bands at 1735 cm−1 attributed to the carboxyl groups in PEUS and by the appearance of bands at 1050 cm−1 which indicates the presence of an apatite phosphate layer on the surface of material as displayed in Figure 9(a). The X-ray diffraction spectra pattern displayed in Figure 9(b) showed a maximum increase at approximately 31° in 2θ after 7 days of soaking, which confirms the growing of the apatite-like phase. The SEM image displayed in Figure 9(c) allows observing the surface morphology change with the soaking time. A layer of spherical particles covering the material surface can be observed, checking the presence of apatite in the surface of the materials.
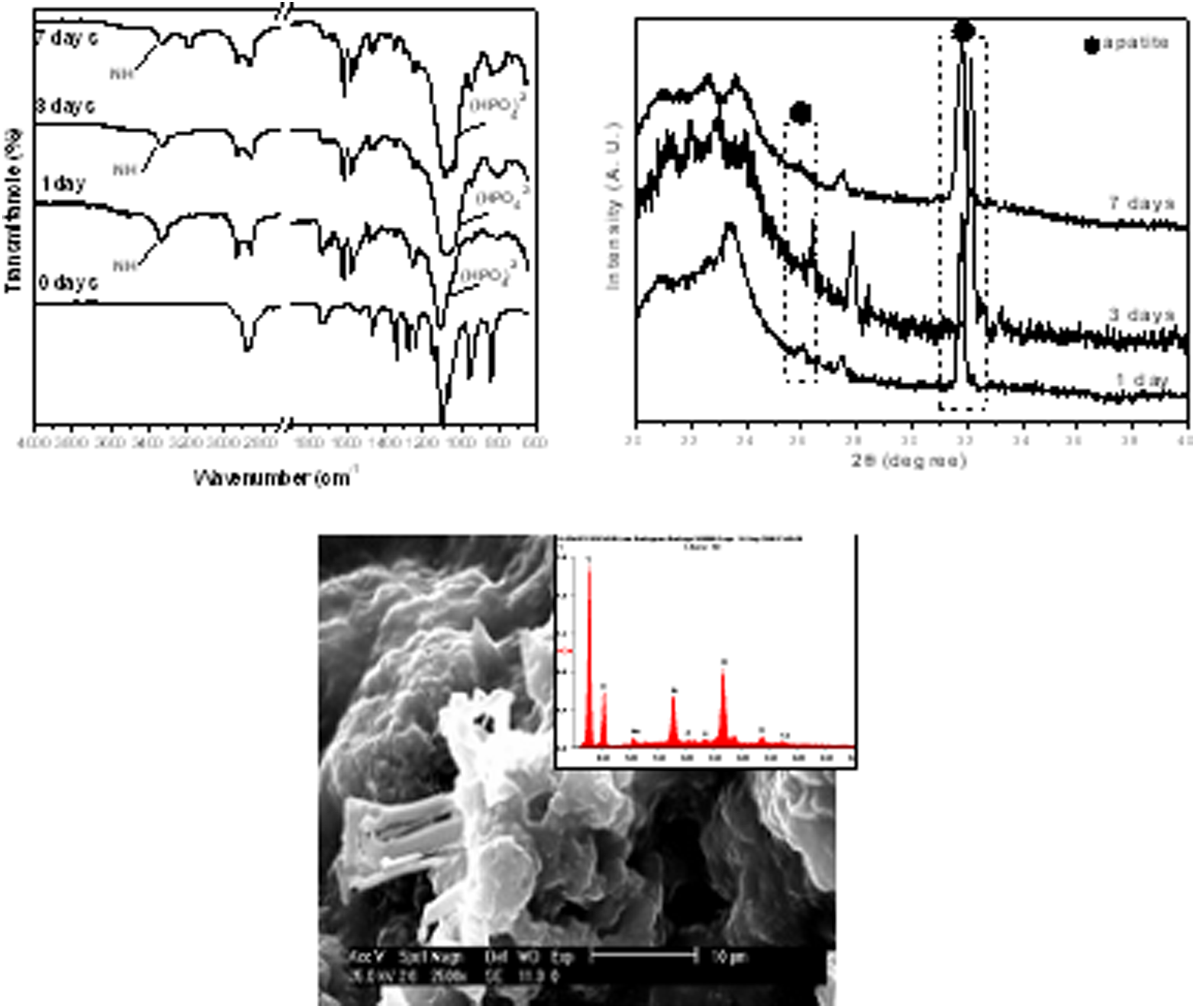
ATR-FTIR, WAXD, and SEM of PEUS1500 after soaking in SBF at different times. ATR-FTIR: attenuated total reflectance-Fourier transformed infrared; WAXD: wide-angle X-ray diffraction.
This behavior was similar to the obtained results from the cytotoxicity assay (MTT assay) where the material was tested for 1, 2, and 7 days. In Figure 10, it can be see that the cellular proliferation for PEUS1500 was not observed in the first day and was low for PEUS3500. In the second day, there is a significant increase in the cell proliferation for both materials and was about 80% for the 7 days. From these results, we can say that materials are not cytotoxic. The cytotoxicity at the first day is high due to the acidity of material as mentioned earlier. From the second day, the acidity decreases as observed in Figure 8 and the solution is less acidic, which increases the cell proliferation.
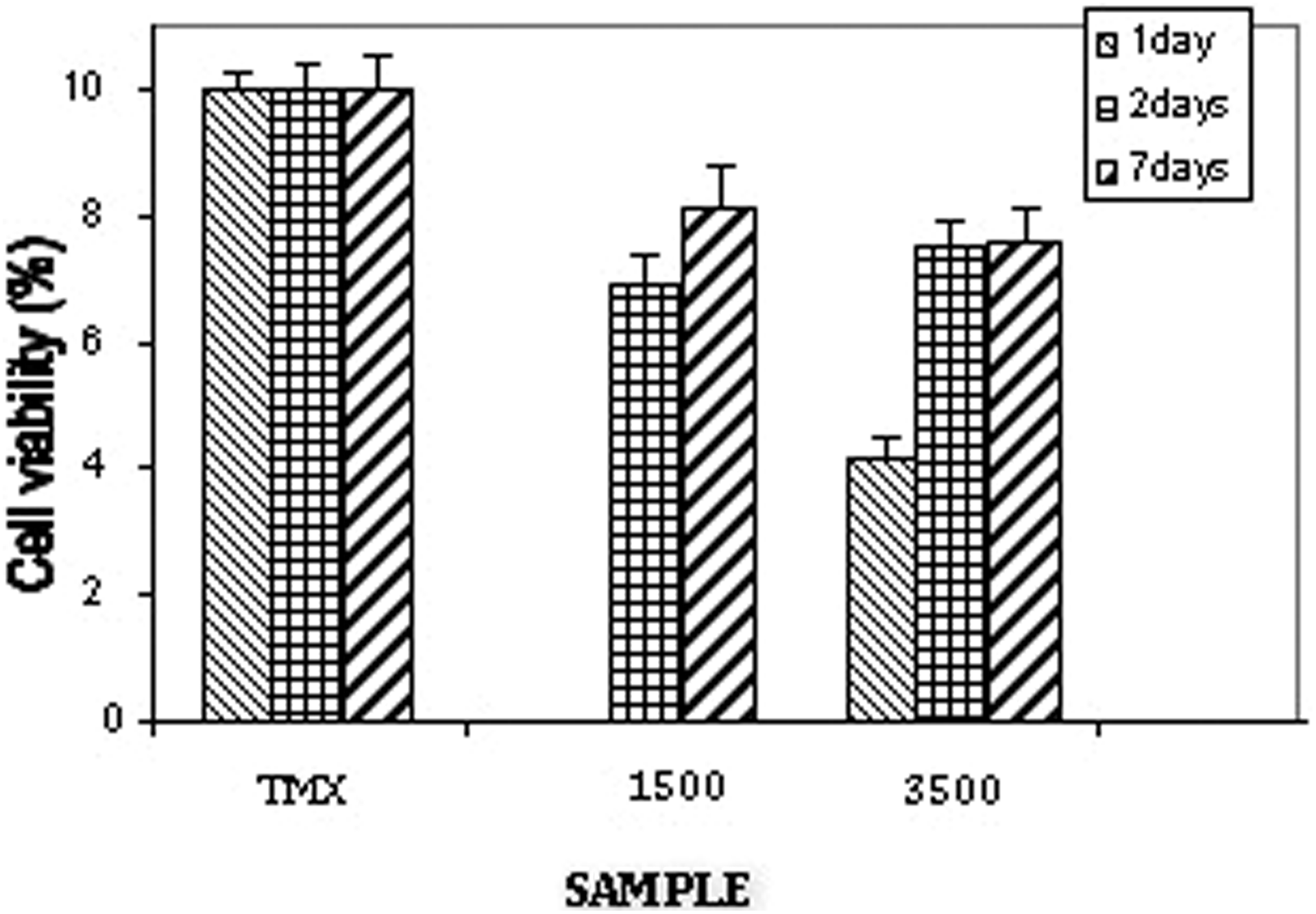
MTT assay for PEUS1500 and PEUS3500. MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
In this work, the use of nanosilica particles in degradable polyurethane matrices is proposed to prepare osteopromoting materials. The presence of hydroxyl groups on the surface of the nanosilica particles must induce the nucleation of apatite, which is an osteoconductive material, while modifications in the polyurethane structure should allow the tailoring of the degradability rate and besides ensuring the material bioabsorbability, without being cytotoxic.
Conclusions
Polyesters and polyurethane/silica composites were successfully synthesized, and they showed fast hydrolytic degradation due to the interaction between the silica and the polyesters chain of the PEUS, however, are not cytotoxic materials. Composites represent a family of scaffolds with potential application in bone tissue engineering. However, a full evaluation of the cytocompatibility, in vitro cell culture studies, and in vivo cell culture investigations should be the focus of future studies to continue the scaffold developments.
Footnotes
Acknowledgment
The authors would like to thank CONACYT (México) and IPN (México) for the financial support.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by CONACYT (México) and IPN (México) and by CICYT MAT2007-63355 (Spain) project.