
Abstract
Two diblock copolymers of poly(methyl methacrylate)-block-poly(styrene) with chlorine as terminal group (PMMA-b-PS-Cl) were synthesized via two-step atom transfer radical polymerization. The structures of the block copolymers were characterized by Fourier transform infrared spectroscopy, proton nuclear magnetic resonance, and gel permeation chromatography. Thermal properties including glass transition temperature (Tg) and thermal stability were studied by differential scanning calorimetry and thermogravimetric analysis (TGA), respectively. The block copolymers of PMMA-b-PS-Cl exhibited two glass transitions, which were attributed to the Tgs of PMMA and PS segments, respectively. According to TGA, thermal decompositions of PMMA macro-initiator and PMMA-b-PS-Cl block copolymers had two stages. The weight loss ratio in the second stage was more significant than that in the first stage, which may be attributed to the separation of the halogen atom from the terminal group and the formation of a double bond. The breaking down of the backbone dominates in the second stage in which the weight loss ratio was more than 70%, represented the main stage of pyrolysis. It was found that the introduction of the PS chain remarkably enhanced the thermal stability of the copolymer, thus endowing the block copolymers high activation energy for thermal decomposition. On the other hand, the remaining two pyrolysis procedures further indicated that thermodynamic mechanism didn’t change due to the introduction of PS segments.
Introduction
Block copolymers with different sequence structures from different monomers have various distinct properties, 1 –5 which not only give some particular use in polymer modification but also have important significance on development of novel materials. Meanwhile, they provide many important models for the study on polymer physics. The block copolymers usually presents not only excellent performances but also complex composition, structure, and morphology, which can be useful in a variety of applications such as thermoplastic elastomers, foams, and adhesives. 6 –9 Over the last decades, copolymerization has been realized as effective approaches to improve physical and mechanical properties of polymeric materials. 10,11
Block copolymers can form meso structures at the nanometer-length scale, which has been the subject of intense research for the past four decades. Recently, constructing block copolymers with hierarchical structures has been the new focus of research.
12
–14
With the application of these block copolymers with hierarchical structures and special morphology in functional material fields, the thermal stability of block copolymers remains an important aspect that should be considered seriously. The enhancement of thermal stability of polymers has received attention from academic and practical points of view. Intensive efforts have been made to understand the mechanism of the degradation of polymers and to design stabilizers having high inherent chemical efficiency. Maurizio Penco et al.
15
studied the thermal properties of multi-block copolymers on the basis of poly(
The thermal properties of block copolymers are widely studied in the decades. 17 –24 However, the relationship of block ratios of different block copolymers on thermal decomposition has seldom been reported. Poly(styrene) (PS) and poly(methyl methacrylate) (PMMA) are classical polymers and given more interest in polymer science. 25,26 They have similar glass transition temperatures (Tgs) and are immiscible, except in the case of small alkyl groups and at low molecular weight. 27,28 The synthesis of PMMA-b-PS has industrial advantages with its wide coverage of Tgs. 29,30 In this article, the effect of block ratios on the thermal behavior and thermal decomposition kinetics of PMMA-b-PS has been initially investigated.
Experiment
Materials and purification
The monomers, styrene (St) and methyl methacrylate (MMA), were purchased from Tianjin Fuchen Chemical Reagents Factory (China). MMA was washed three times with 5% sodium hydroxide solution to remove the inhibitor and then with distilled water until the monomer became neutral. A certain amount of magnesium sulfate was subsequently added to the solution after 24 h and then the solution was vacuum distilled at 60°C. The treatment of St was similar to that of MMA. However, the final vacuum distillation was applied at 80°C, and copper (I) chloride (CuCl) into glacial acetic acid was stirred without light for 10 h. The obtained solution was washed with ethanol and acetone until the solution turned gray–white. The prepared CuCl powder was placed in a brown bottle after heating for 24 h under vacuum at 40°C. Tetrahydrofuran (THF) was distilled under atmospheric pressure at 70°C after reflux using sodium. Dimethyl formamide (DMF) was employed after vacuum distillation at 90°C. Pentamethyldiethylenetriamine (PMDETA) and ethyl 2-bromoisobutyrate (EBiB) were purchased and used as received from Aladdin Reagent Co., Ltd (China).
Synthesis of PMMA macro-initiator and the block copolymers
PMMA macro-initiator with halogen atoms as the terminal group (PMMA-Cl) was synthesized by atom transfer radical polymerization (ATRP) firstly. The reacting compounds, namely MMA (21.21 mL, 200 mmol), EBiB (0.29 mL, 2 mmol), CuCl (0.20 g, 2 mmol), and PMDETA (0.84 mL, 4 mmol), were dissolved in THF (42.42 mL). The reaction of the compounds was carried out for 5 h in a three-necked flask under nitrogen (N2) atmosphere at 60°C. After the reaction, the mixture was diluted threefold with THF, expelled into an alumina column, and precipitated into methanol. The resultant polymer was filtered and dried overnight under vacuum at 40°C.
Secondly, the block copolymers of PMMA-b-PS with the halogen atoms as the terminal group (PMMA-b-PS-Cl) were synthesized through ATRP using PMMA-Cl as macro-initiator. St (2.23 mL, 20 mmol) was then initiated by the PMMA macro-initiator (2.00 g) with CuCl (0.04 g, 0.4 mmol)/PMDETA (0.17 mL, 0.8 mmol) as catalyst and DMF (10 mL) as solvent. The mixture was continuously stirred at 90°C for 10 h under N2 atmosphere. After the reaction, post-processing was conducted using the previously mentioned method. The block chain length of the copolymer can be changed by altering the molecular weight of PS. Another block copolymer was synthesized when 6.70 mL (60 mmol) St was added. The two kinds of block polymers were named PMMA-b-1PS and PMMA-b-1.8PS based on the actual mole ratio of PMMA and PS in the polymers, which is calculated based on the data of gel permeation chromatography (GPC).
Characterization
Fourier transform infrared (FTIR) spectroscopy was acquired at room temperature by using the WQF-310 FTIR system connected to a computer. The derived spectra ranged from 600 cm−1 to 4000 cm−1. High-resolution proton nuclear magnetic resonance (1H NMR) measurements were performed using a Bruker AV500 spectrometer (Billerica, Massachusetts, USA) with tetramethylsilane as an internal reference. Deuterated chloroform (CDCl3) was used as the solvent for all the samples for NMR measurement. The average molecular weight and molecular weight distributions of PMMA and the block copolymers were determined by GPC calibration, with standard PS and THF as eluent by using Waters 1515 gel permeation chromatograph (Milford, Massachusetts, USA). The eluting rate was 1 mL min−1.
Differential scanning calorimetry (DSC) experiments were performed using 2910MDSC V4.4E from TA Instruments (New Castle, Delaware, USA), equipped with an autosampler and subjected to a heating rate of 10°C min−1 and at a temperature ranging from 25°C to 150°C under N2 atmosphere.
Thermogravimetric analysis (TGA) was conducted using Netzch STA thermogravimetric analyzer (Germany) under N2 atmosphere at four heating rates: 10, 15, 20, and 30°C min−1. TGA curves were obtained under argon atmosphere at a thermal decomposition temperature range from 20°C to 500°C.
Results and discussion
FTIR, 1H NMR, and GPC analyses of polymers
The PMMA macro-initiator and the block copolymers are characterized by FTIR, 1H NMR, and GPC analyses, and are listed in Figure 1, Figure 2, and Table 1, respectively. Figure 1 shows the FTIR of the polymers that the absorption bands at 1731 and 3025 cm−1 are assigned to the –C=O and to the symmetric and asymmetric –CH3 stretching vibrations of PMMA, respectively. The peaks at 1493 and 1600 cm−1 are assigned to the stretching vibration of the benzene ring, whereas the band at 700 cm−1 corresponds to the characteristic absorption of monosubstituted benzene. Peaks disappeared at 1671 and 1636 cm−1, indicating the absence of monomers in the products.
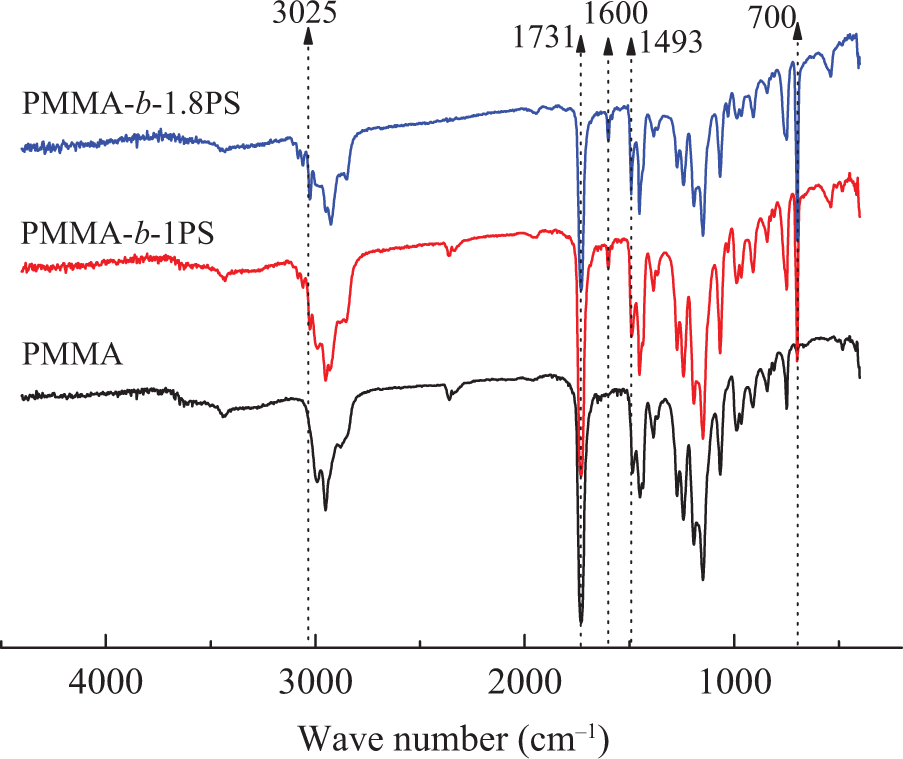
FTIR spectra of PMMA and two block copolymers. FTIR: Fourier transform infrared; PMMA: poly(methyl methacrylate).
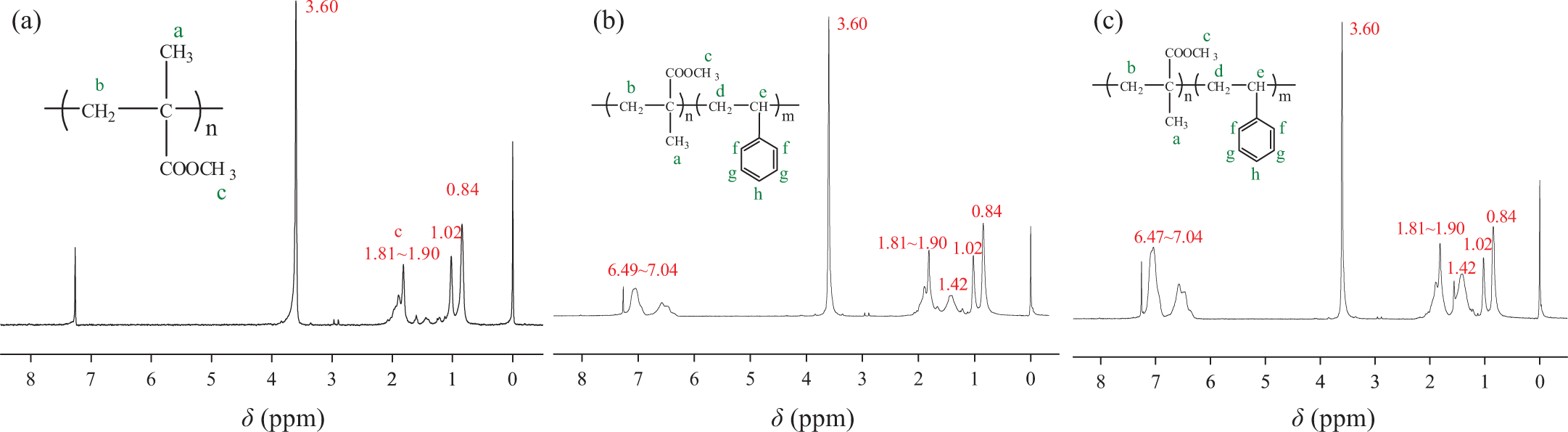
1H NMR spectra of (a) PMMA, (b) PMMA-b-1PS, and (c) PMMA-b-1.8PS. 1H NMR: proton nuclear magnetic resonance; PMMA: poly(methyl methacrylate); PMMA-b-PS: poly(methyl methacrylate)-block-poly(styrene).
Molecular weight and polydispersity results of PMMA macro-initiator and PMMA-b-PS.
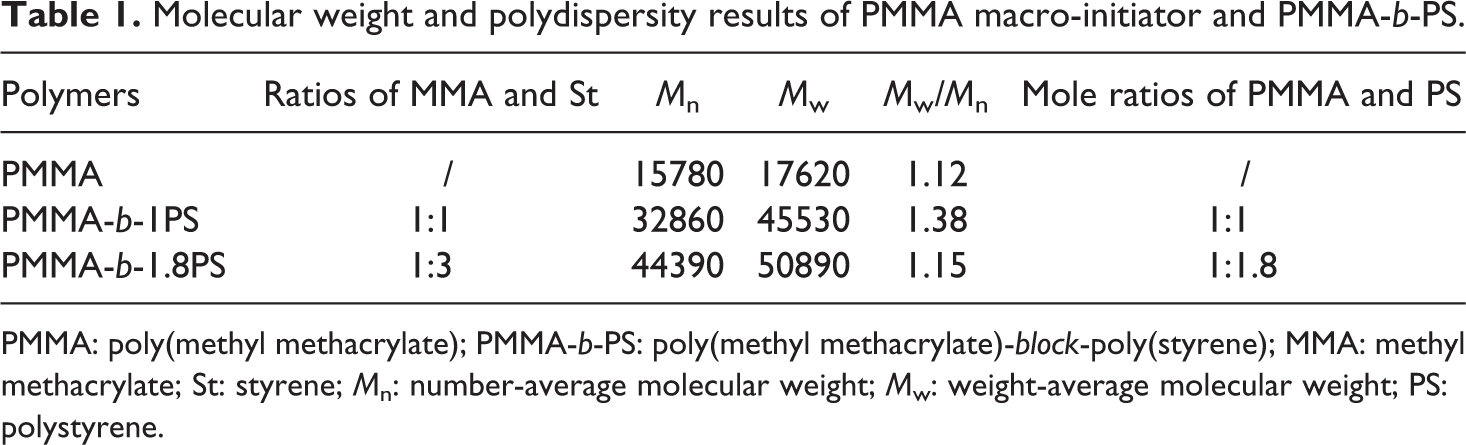
PMMA: poly(methyl methacrylate); PMMA-b-PS: poly(methyl methacrylate)-block-poly(styrene); MMA: methyl methacrylate; St: styrene; Mn: number-average molecular weight; Mw: weight-average molecular weight; PS: polystyrene.
1H NMR spectra of PMMA macro-initiator in CDCl3 and the attributes of the peaks are presented in Figure 2(a). The characteristic peak at 3.60 ppm is clearly attributed to the methyl ester group of PMMA. The multiple peaks at regions ranging from 1.81 to 1.90 ppm and from 0.84 to 1.02 ppm are attributed to the methylene and methyl group of atactic PMMA, respectively. The 1H NMR spectra of the block copolymers are illustrated in Figure 2(b) and (c). The appearance of multiple peaks in the regions ranging from 6.47 to 7.04 ppm confirmed the presence of benzyl protons. Signals originating from the methylene protons of PS were discerned at approximately 1.42 ppm. 26
The molecular weight and polydispersity of the PMMA macro-initiator and the block copolymers are measured by GPC, and the results are illustrated in Table 1.
The mole ratios of PMMA and PS are shown in Table 1, which are calculated by the results of GPC and 1H NMR. The molecular weight distribution of PMMA-Cl is narrow, which is a typical characteristic of ATRP. In the case of the block copolymers, the molecular weight distribution increases, especially at the lower addition of St compared with that of PMMA-Cl, which is caused by fast initiation, slow propagation, and nontermination of ATRP progress. When a small amount of St is added, PMMA-Cl rapidly conducts the initiation of St and the reaction terminates rapidly, which introduce a wide distribution of molecular weight of block copolymer. By increasing the St feeding ratio, the percentage of St conversion is depressed, but the distribution remains narrow. Then, we could obtain the different mole ratios of PMMA to PS, 1:1 and 1:1.8. In the following context, the two block copolymers were named as PMMA-b-1PS and PMMA-b-1.8PS.
DSC analysis
The Tg of the three polymers are measured by DSC as shown in Figure 3. It is found that the PMMA macro-initiator indicated only one Tg, whereas PMMA-b-1PS and PMMA-b-1.8PS indicate two Tgs, conforming to two discontinuous separate micro-phases behavior in block copolymer. The lower Tg is assigned to the PS segment and the higher Tg is to the PMMA segment. Figure 3 shows that each Tg of the PMMA segment in the copolymer is higher than that of the pure PMMA macro-initiator, proving the limited mobility of the PMMA segment in the block copolymer because of the introduction of the rigid PS segment. In the DSC curves, no melting peak of the crystal is observed within the indicated temperature range, denoting that the block copolymers are completely amorphous.
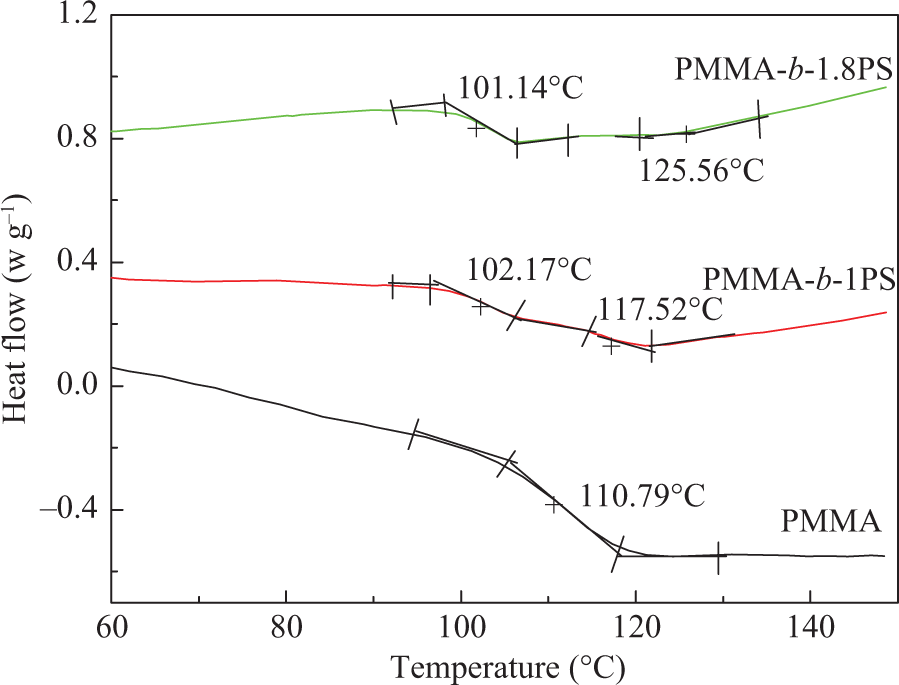
DSC curves of PMMA-Cl, PMMA-b-1PS, and PMMA-b-1.8PS. DSC: differential scanning calorimetry; PMMA-Cl: polymethyl methacrylate with chlorine initiator; PMMA-b-PS: poly(methyl methacrylate)-block-poly(styrene).
Thermal stability and thermal decomposition kinetics of the copolymer
To investigate the thermal stability of the copolymers, these polymers are examined by TGA. 31 The TGA and differential TG curves of PMMA, PMMA-b-1PS, and PMMA-b-1.8PS at four different heating rates of 10, 15, 20, and 30°C min−1 are shown in Figures 4, 5, and 6, respectively.
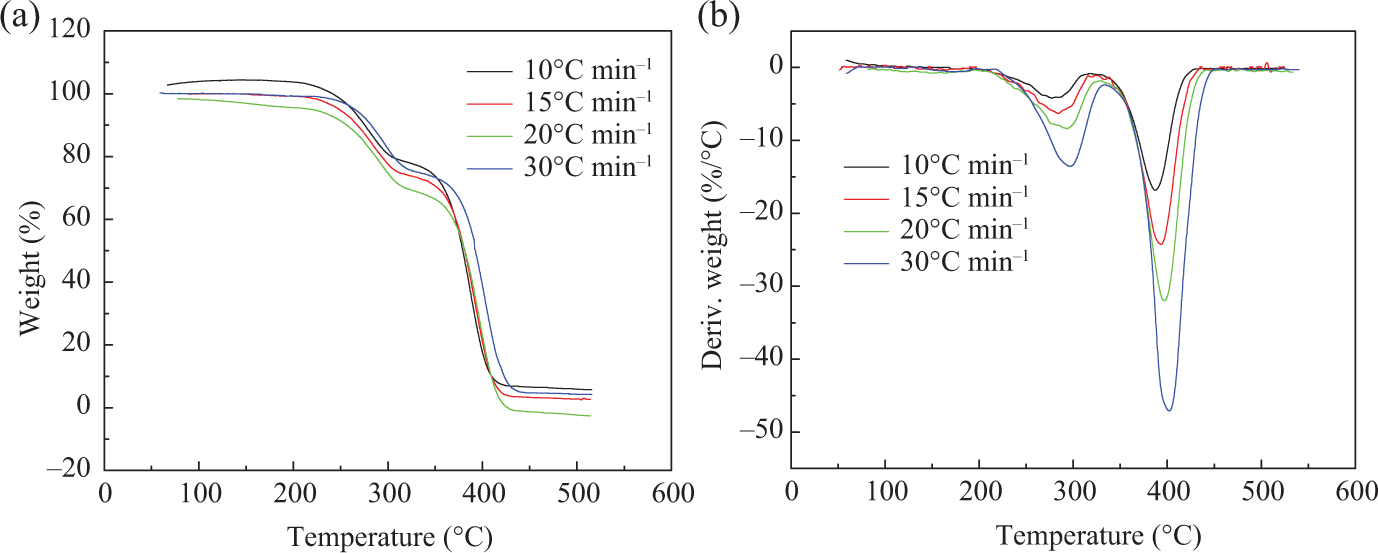
(a) TGA and (b) DTG curves of PMMA. TGA: thermogravimetric analysis; DTG: differential thermogravimetric.
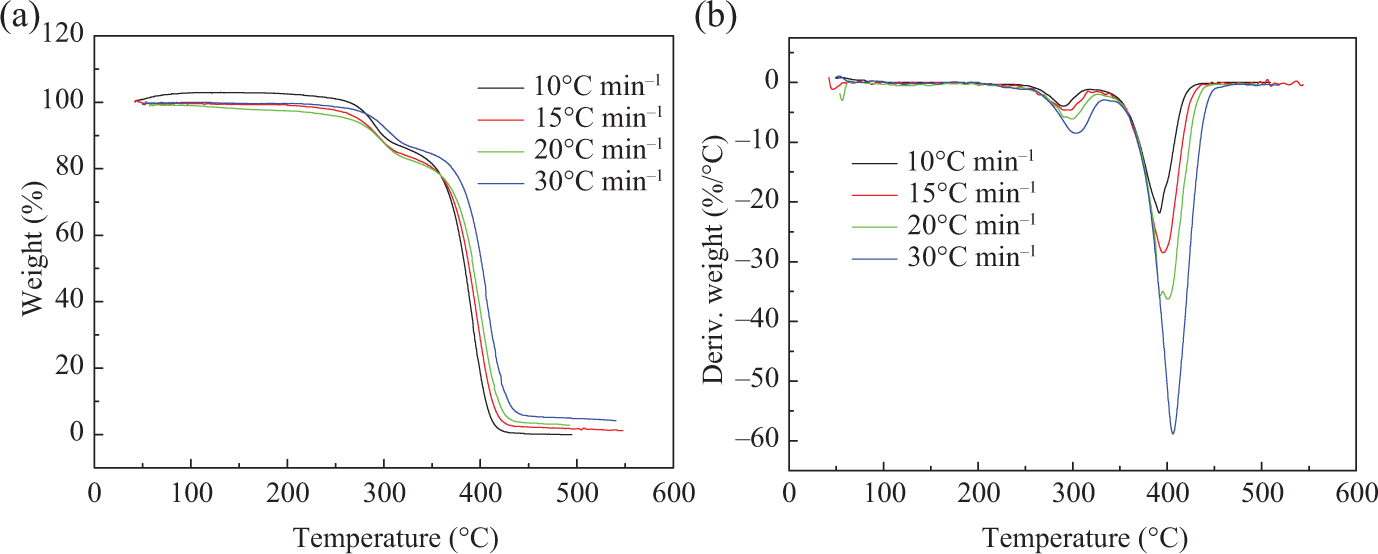
(a) TGA and (b) DTG curves of the block copolymer PMMA-b-1PS. TGA: thermogravimetric analysis; DTG: differential thermogravimetric; PMMA-b-PS: poly(methyl methacrylate)-block-poly(styrene).
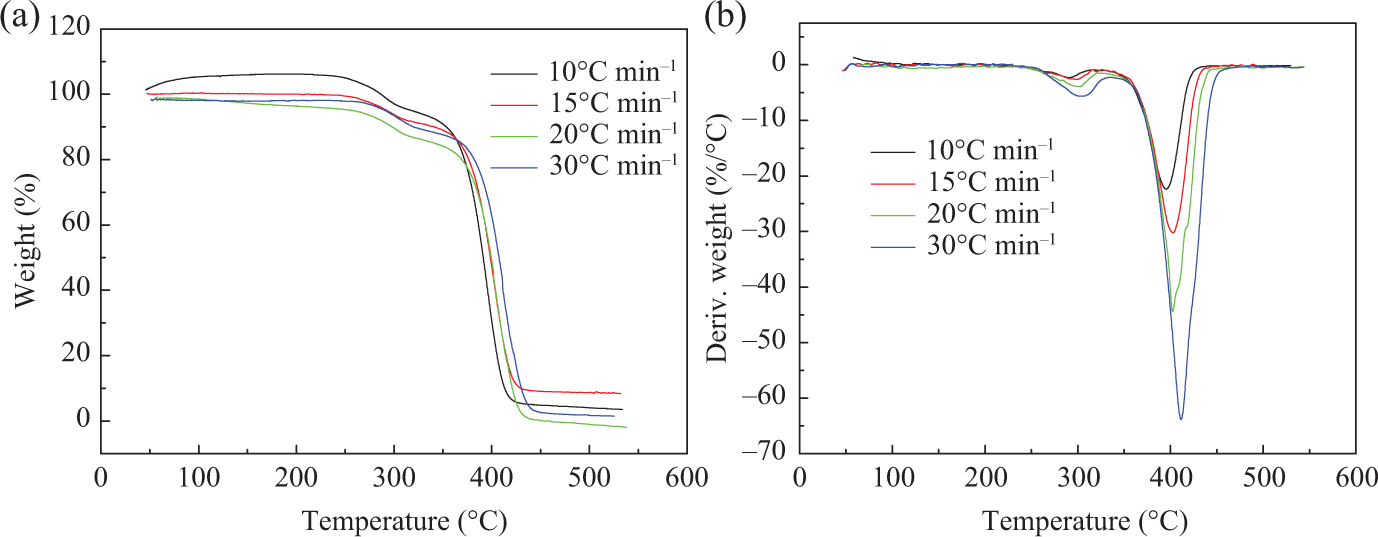
(a) TGA and (b) DTG curves of the block copolymer PMMA-b-1.8PS. TGA: thermogravimetric analysis; DTG: differential thermogravimetric; PMMA-b-PS: poly(methyl methacrylate)-block-poly(styrene).
Decomposition temperatures at the maximum rate of decomposition (Td, max) are listed in Table 2.
Maximum pyrolysis rate of temperatures T1 and T2 at different heating rates.
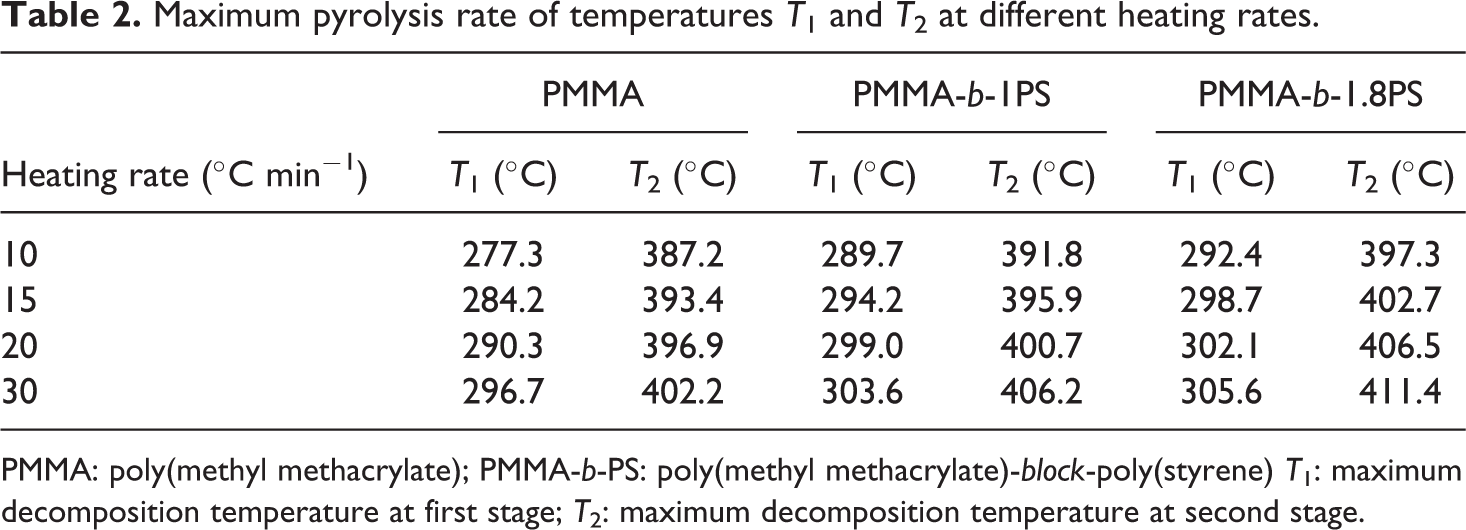
PMMA: poly(methyl methacrylate); PMMA-b-PS: poly(methyl methacrylate)-block-poly(styrene) T1: maximum decomposition temperature at first stage; T2: maximum decomposition temperature at second stage.
These results show that all samples, that is, PMMA-Cl and the two copolymers, have two Td, max during the thermal decomposition process, T1 and T2 (T1 and T2 correspond to the first and second decomposition temperatures at the maximum rate of decomposition, respectively), which isn’t relevant to the addition of PS block. It is reported that the polymers synthesized by ATRP methods usually end with the halogen terminal group, which easily initiate breakdown of backbone during heating. Thus, it may have two decomposition processes. The pyrolysis reactions often start with the halogen terminal group and then mainly attribute to chain depolymerization. 32,33
As shown in Table 2, it is found that the decomposition temperatures increase as the heating rate increases. Both T1 and T2 increase successively in the order of PMMA, PMMA-b-1PS, and PMMA-b-1.8PS at the same heating rate. Therefore, Td, max increases with the increase of the PS segments, which means that the improvement of thermal stability is mainly attributed to the introduction of rigid PS segments into polymer backbone. 34,35
On the basis of the above analysis, pyrolysis process of all polymers can be divided into two stages. The first stage has maximum decomposition temperature T1 ranging from 200°C to 340°C, during which the mass loss of copolymer is not significant, and the mass loss of PMMA-b-1PS is 3% (14% to 17%) and PMMA-b-1.8PS is about 8% (5% to 13%), while the PMMA initiator is about 9% (22% to 31%). According to the previous study on the thermal decomposition of polymers synthesized by ATRP, 36 the main pyrolysis products in this stage should include monomers and oligomers initiated by terminal Cl. The second stage has maximum decomposition temperature T2 ranging from 340°C to 470°C, which is considered to be the principal pyrolysis stage of backbone. In this stage, significant weight loss occurs, and weight loss reaches above 70%.
In order to investigate the pyrolysis behavior of PMMA-b-PS, thermal decomposition kinetics parameters are fitted using the Kissinger equation. The activation energies of two pyrolysis stages are estimated, and the decomposition mechanism of the pyrolysis reaction is determined.
Multiple scanning rate methods, such as the Kissinger method, 37 utilize multiple TGA curves measured at different heating rates for dynamic analysis. A more reliable activation energy (E) can be acquired using the Kissinger method, without involving the dynamics model function as a premise. The Kissinger equation is shown as follows 38 –40 :
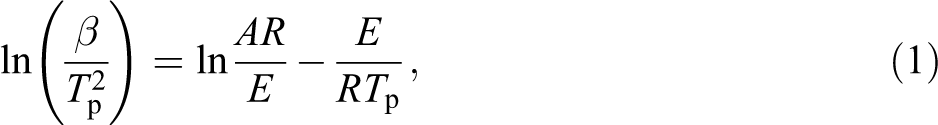
where β represents the heating rate (°C min−1), Tp is the maximum pyrolysis temperature rate (K), A is the exponential factor (min−1), R is the ideal gas constant (8.314 J mol−1 K−1), and E is the activation energy (kJ mol−1).
Equation (1) emphasizes that
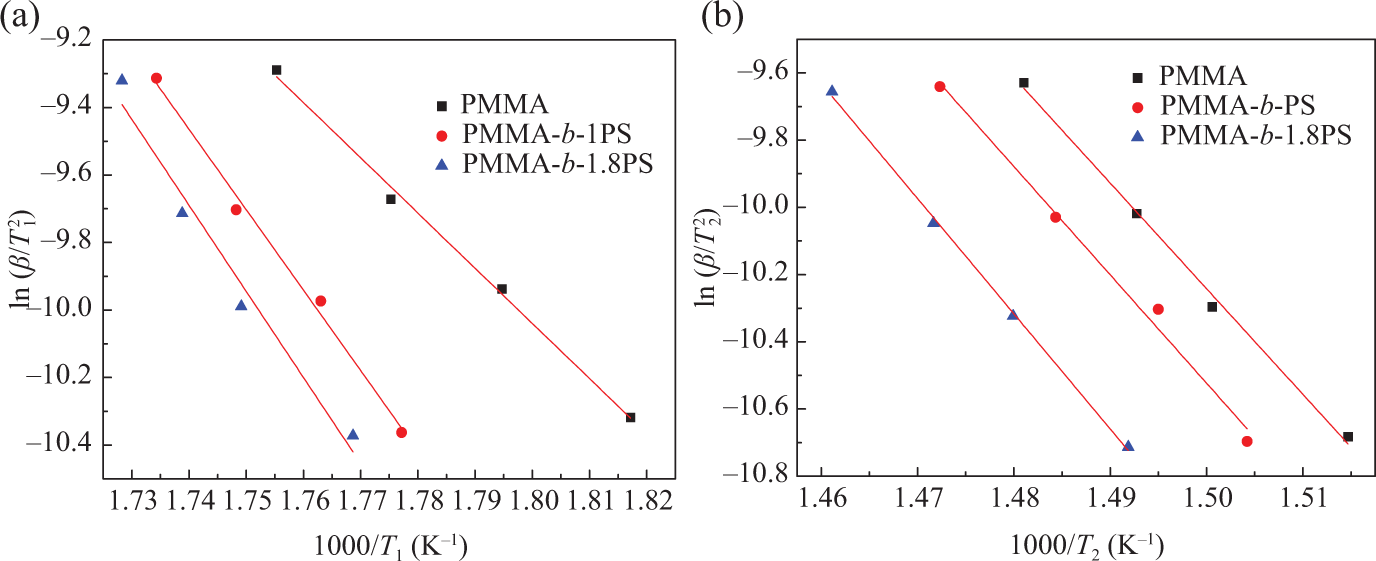
The Kissinger method fitting chart of (a) the first stage and (b) the second stage.
Linear regression curves and activation energy of three polymers during two thermal decomposition stages.
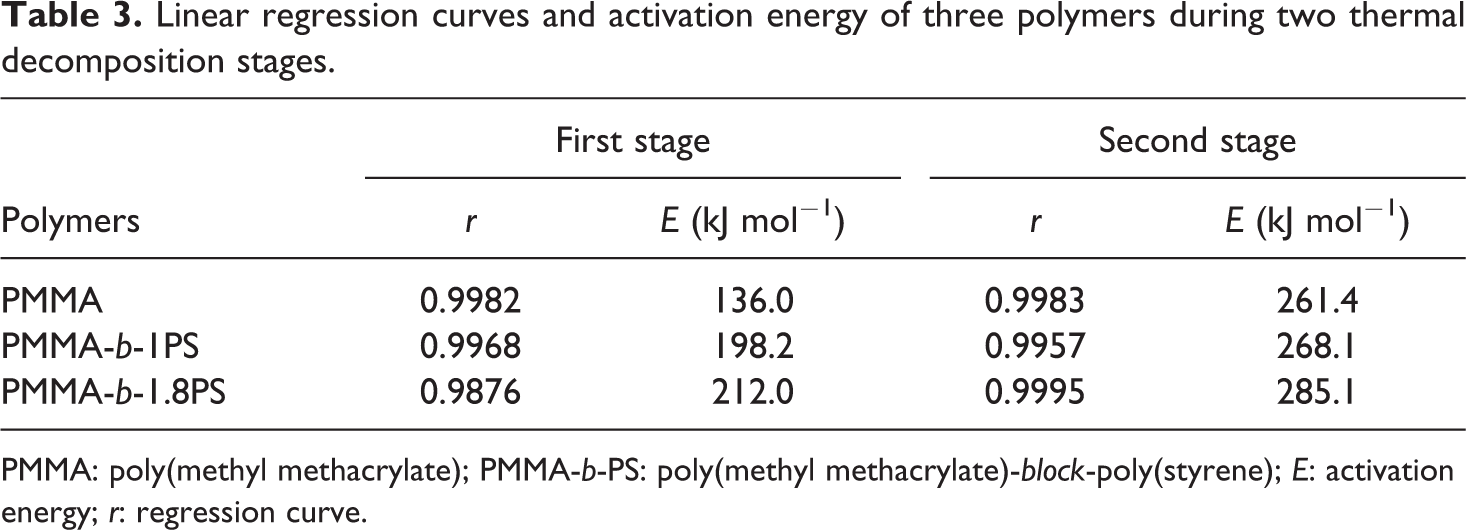
PMMA: poly(methyl methacrylate); PMMA-b-PS: poly(methyl methacrylate)-block-poly(styrene); E: activation energy; r: regression curve.
Here r represents linear correlation. It is found that the pyrolysis stages activation energy of the copolymer increased with the addition of PS block to copolymer backbone. Moreover, the longer the PS block is, the higher the activation energy of the pyrolysis decomposition. 41 Meanwhile, the activation energy at the second stage is higher than that of first stage, which may due to the different mechanism of pyrolysis of two stages.
The integral method is often used to acquire the order of reaction. Therefore, for the first-order reaction, we used the following integral form dynamics function:

The following Coats–Redfern method is then employed:
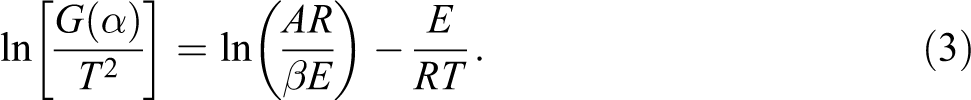
In most cases, the majority of thermal decomposition reactions can be assumed as the first order of reaction, that is, n = 1. Following the Coats–Redfern approach, equation (2) is integrated into equation (3), thereby obtaining equation (4), which is shown as follows:
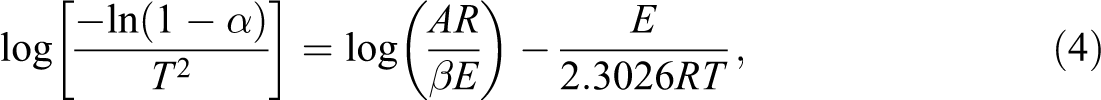
where α is the weight loss ratio, β is the heating rate (°C min−1), T is the temperature (K) at certain conversion point (α), A is the exponential factor (min−1), R is the ideal gas constant (8.314 J mol−1 K−1), and E is the activation energy (kJ mol−1).
By deriving
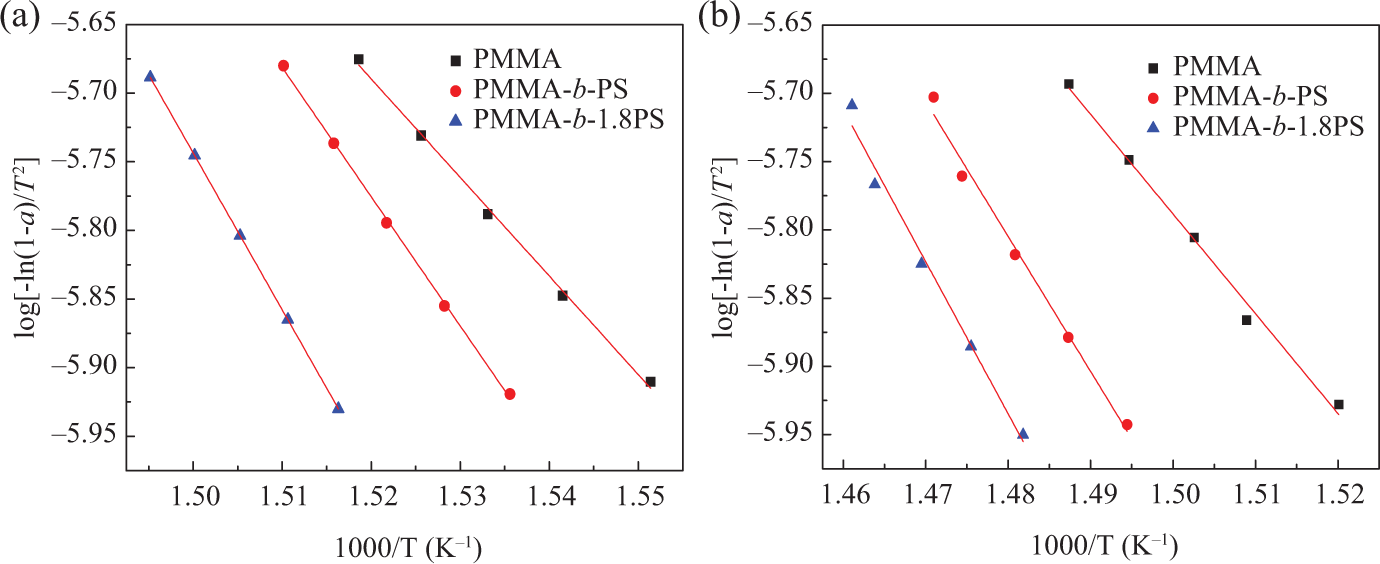
log[−ln (1−α)/T2]∼1/T at different heating rates: (a) β =10°C min−1 and (b) β = 30°C min−1.
The weight loss ratios are determined from the TGA data under an argon atmosphere. The linear correlations after linear regression are presented in Table 4.
Linear regression curve and apparent activation energy at different heating rates.
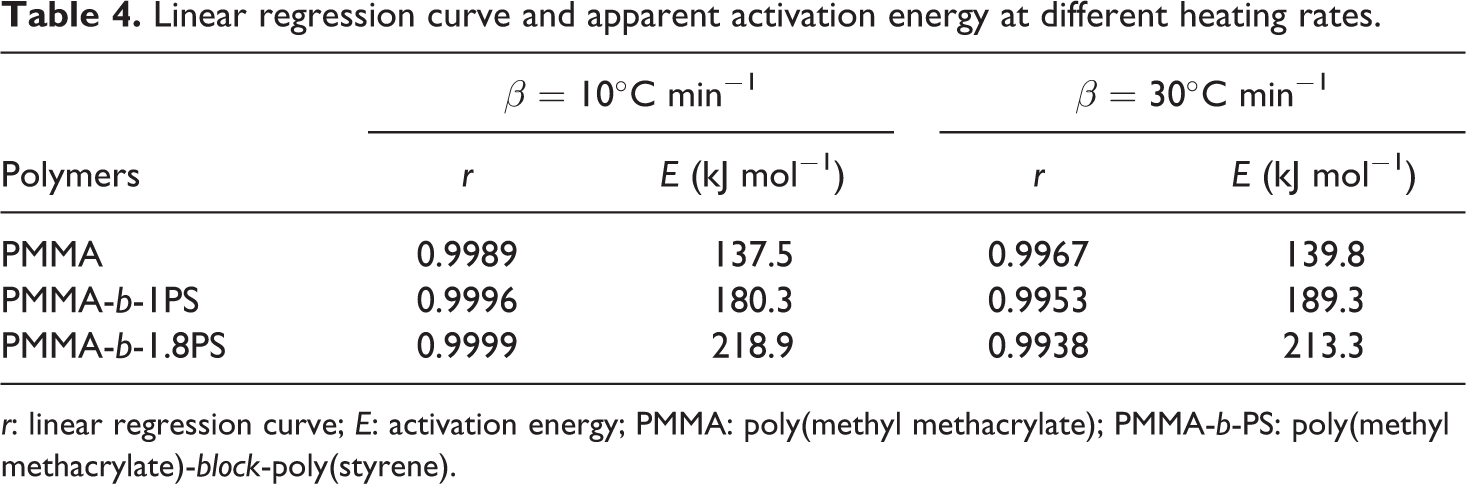
r: linear regression curve; E: activation energy; PMMA: poly(methyl methacrylate); PMMA-b-PS: poly(methyl methacrylate)-block-poly(styrene).
We can obtain excellent fitting linear curves at higher heating rates, demonstrating that the reaction types of thermal decomposition belong to the first-order reaction during decomposition under an argon atmosphere. By comparing the activation energy data listed in Table 4, it is found that the activation energies of the same polymer are close to each other at different heating rates. It proves the first-order reaction prediction as discussed earlier. But at the same heating rate, the decomposition activation energies increase by order of PMMA, PMMA-b-1PS, and PMMA-b-1.8PS, which means that introducing the PS segment on PMMA can improve the polymer thermal stability.
Based on the thermodynamic kinetics study, we could summarize the mechanism of pyrolysis process of different polymers. The thermal decomposition processes are shown in Figures 9 and 10.
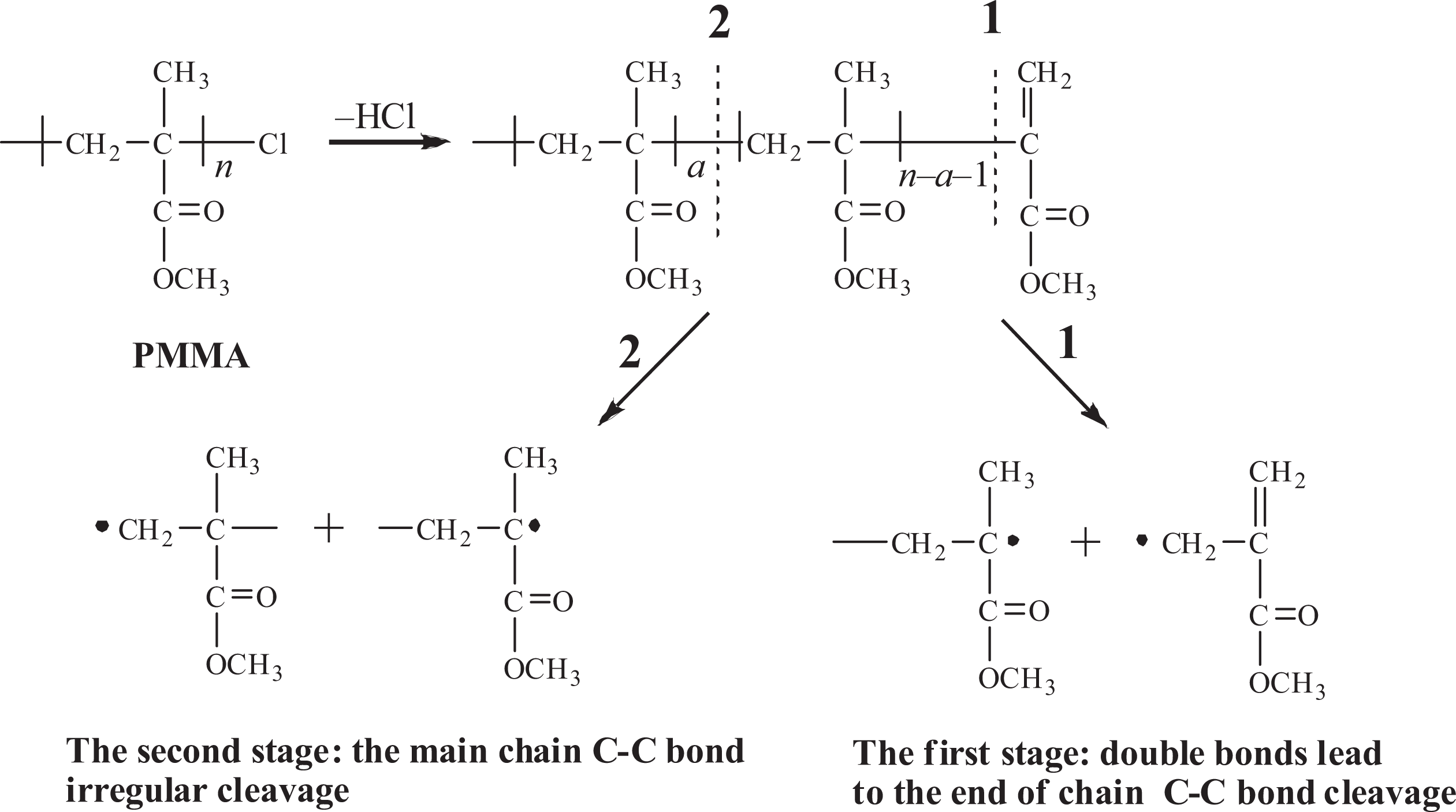
Thermal decomposition reaction of PMMA. PMMA: poly(methyl methacrylate).
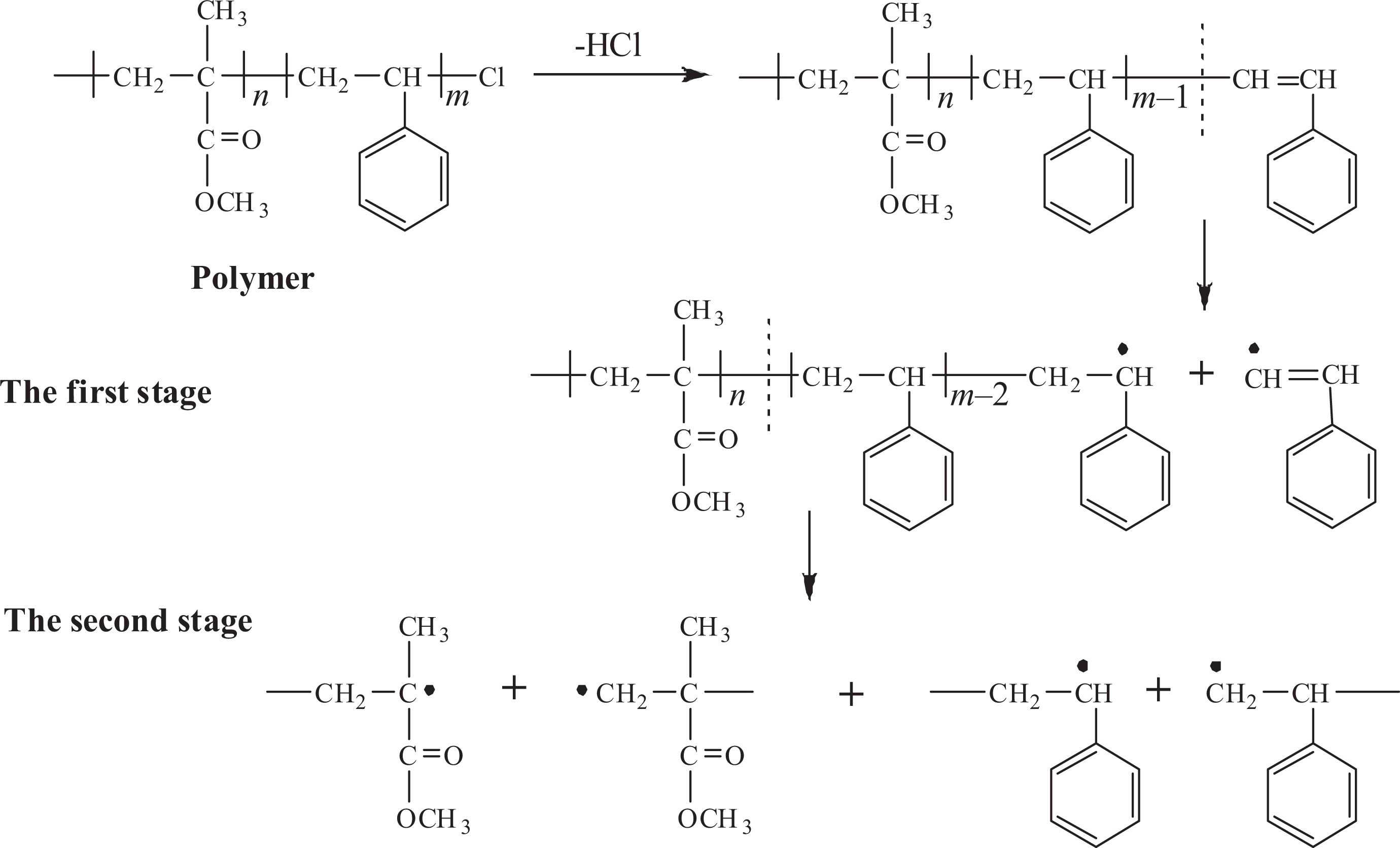
Thermal decomposition of the block copolymers.
As shown in Figure 9, it could utilize a two-step decomposition procedure for describing PMMA-Cl pyrolysis process, C–Cl bond is first broken accompanied by C=C bond formation with a lower activation energy and low pyrolysis temperature. 42 As the temperature is increased, it is facilitated that the main-chain degradation exhibits higher activation energy accompanied by the release of hydrochloric acid. 43
Similar to PMMA-Cl pyrolysis process, the copolymer still shows a two-step pyrolysis process. The first pyrolysis step initiated by terminal Cl group exhibits higher activation energy compared to that of PMMA-Cl, which suggests that first pyrolysis step of copolymer is getting harder due to the addition of PS segment. Meanwhile, the second pyrolysis step mainly attributes to PMMA and PS main backbone breakdown at the same time since it proves to be first-order reaction and no detection of third pyrolysis process by thermodynamic study. During second pyrolysis, first the long backbone decomposed to shorter chains, followed by the release of volatile low molecular dimmer or trimer as well. 44 Table 4 shows the introduction of the PS chain endowing the block copolymer high activation energy for thermal decomposition, thus remarkably enhancing the thermal stability of the copolymer. But the remaining two pyrolysis procedures further indicate that thermodynamic mechanism doesn’t change due to the introduction of PS segments.
Conclusions
Two diblock copolymers of PMMA-b-PS-Cl with different block ratios were synthesized via two-step ATRP. The structures were characterized by FTIR, 1H NMR, and GPC analyses, and the thermal properties were investigated by DSC and TGA.
From the DSC analysis, it is found that the block copolymers exhibited two Tgs. According to TGA, both the PMMA macro-initiator and the block copolymers have two thermal decomposition stages. The weight loss ratio in the second stage is more than that in the first stage, which may be caused due to the breakage of the halogen atom in the terminal group and the formation of a double bond that leads to C–C fracture. It is shown that the Td, max increases with the increase of the PS segments, which means that the improvement of thermal stability is mainly attributed to the introduction of rigid PS segments into polymer backbone. Finally, the thermal decomposition kinetics is investigated and found that the pyrolysis stages activation energy of the copolymer increased with the addition of PS block to the copolymer backbone. Moreover, the longer the PS block is, the higher the activation energy of the pyrolysis decomposition. Meanwhile, activation energy at the second stage is higher than that of the first stage, and the activation energies of the same polymer are close to each other at different heating rates. Both the PMMA macro-initiator and block copolymers exhibit two-step decomposition procedure, and the two pyrolysis procedures indicate that thermodynamic mechanism doesn’t change due to the introduction of PS segments.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors are grateful for the support from the Project of National Natural Science Foundation of China (no. 51103117), National Natural Science Foundation of Shaanxi Province (nos 2013JQ2010 and 2013JM2012), NPU Fundamental Research Foundation (nos JC201158 and 3102014JCQ01089), and Graduate Starting Seed Fund of NPU (no. Z2011015).