
Abstract
Illnesses with both cognitive and affective symptoms are common in older adults and have a broad neuropsychiatric differential diagnosis. We present a case of a 66-year-old female with fluctuating affective symptoms on a background of chronic dysexecutive cognitive symptoms, posing an intriguing diagnostic challenge and ultimately considered to be probable Kleine-Levin Syndrome (KLS) and cerebellar cognitive affective syndrome (CCAS). To our knowledge, this is the first published instance of both diagnoses not only co-occurring, but having a contemporaneous onset. This is followed by an overview of common differential diagnoses for such presentations, and a detailed narrative review of the features of and KLS and CCAS, highlighting their possible intersection. To account for the manner in which CCAS may have precipitated KLS in this case, we discuss the possible impact of posterior cerebellum damage on thalamic, default mode network, and other otherwise undamaged cerebral areas’ function, via the disruption of broader neuronal connectivity networks.
Keywords
Background
Illnesses with both cognitive and affective symptoms are common in older adults and have a broad neuropsychiatric differential diagnosis. This includes diagnoses of delirium, dementia with behavioural and psychological symptoms, toxidromes (including those associated with prescribed medications), postictal psychosis, and metabolic causes of encephalopathy, in addition to primary psychiatric diagnoses.
Here, we present a case of a 66-year-old female with fluctuating affective symptoms on a background of chronic dysexecutive cognitive symptoms, posing an intriguing diagnostic challenge. Informed, written consent for publication was obtained from her. This is followed by an overview of common differential diagnoses for such presentations and a discussion of the features of both Kleine-Levin Syndrome (KLS) and cerebellar cognitive affective syndrome (CCAS).
Case Presentation
AC, a 66-year-old single, unemployed woman well-supported by children, including 1 with whom she lived, presented to the emergency department following an impulsive overdose of prescription desvenlafaxine and diazepam. She had been prescribed desvenlafaxine for decades, despite an ostensible history of bipolar affective disorder (BPAD). On initial presentation, she was dysphoric and reported longstanding, intermittent, and brief passive suicidal ideation when overwhelmed by interpersonal conflict, as she had been on the day of her presentation – having entered a verbal dispute with her ex-husband. This was her first suicide attempt. Longstanding histories of mild volatility of affect and mild impulsivity were elicited on collateral history, and corroborated cross-sectionally on mental state examination. Despite this, there was no history of impaired interpersonal functioning to support the initial differential diagnosis of borderline personality vulnerabilities.
She was admitted to the inpatient psychiatric unit for 2 days for crisis containment and further assessment. Her dysphoria rapidly ameliorated, her suicidal ideation resolved, and she expressed a preference for outpatient follow-up. Medications at discharge included venlafaxine 150 mg mane, and olanzapine 5 mg nocte, which had been commenced for initial insomnia on her first night on the ward in the context of her reported history of BPAD.
On our review of AC in the community, she continued to report fluctuating, passive suicidality arising from a longstanding feeling of burdening her family. She nonetheless engaged with warmth and was able to guarantee her safety. Notably, beyond her lability, there were no features on mental state examination nor history to support an impression of hypomania, nor any features of a depressive illness. Similarly, there was no evidence of identity diffusion or ‘primitive’ psychological defences that characterize personalities operating at the borderline level. 1 She did not meet any DSM-V personality disorder diagnostic criteria. 2
On further exploration, AC noted recurrent week-long episodes of hypersomnolence, occurring approximately once every few months. She had recovered from one such episode a week before her medication overdose. She also described intense hypersexuality during these episodes.
Medical history was notable for a left acoustic neuroma resection in her 20s due to progressive ataxia. AC described a dramatic personality change immediately following this from being ‘shy’ and ‘meek’ to being cantankerous, impulsive, curious, driven, open to new experiences, and extraverted.
Shortly after the resection, AC was diagnosed with BPAD by a psychiatrist due to the presence of post-procedural and enduring, mild impulsivity, lability, and loquaciousness; and her development of periods of dysphoria and irritability with social withdrawal. During these periods, AC would sleep for 20 hours a day and would experience weight gain from overeating during the brief periods she was awake. She recalled little of these episodes beyond feeling depressed in mood. The episodes lasted for approximately a week before her mental state reverted to her new, garrulous baseline mental state. A trial of lithium resulted in nausea, brain fog and dizziness; AC could not remember the dose she was taking, or if she had been formally diagnosed with lithium toxicity. Although her lability and mild impulsivity were longitudinally noted, they never became functionally debilitating, nor did she develop other features of hypomania. Eventually, she was commenced on desvenlafaxine for her periods of hypersomnolence and depressed mood and remained on this for decades. She did not describe subjective improvement with this treatment, and the frequency, length and intensity of her symptoms were unchanged.
AC did not experience excessive daytime sleepiness nor hypersomnia between these episodes. She described her sleep as refreshing at baseline, and while she had not been pervasively observed through a given night of sleep in recent years, there was no evidence of apnoeic episodes on observation of her sleep overnight by family. She had never experienced episodes consistent with cataplexy nor migraine. There was no other medical or developmental history of note.
Neurological examination revealed ongoing left CN I-XII palsies following her acoustic neuroma resection several decades ago. She had horizontal nystagmus on horizontal gazing. There was a subtle weakness of left-sided upper limb movements relative to the right. Left limb ataxia was apparent. There was no gait or truncal ataxia. She had a positive glabellar tap sign, but no other frontal release signs were elicited.
AC’s cognitive impairments on initial assessment relative to aged-matched normative or expected data.

Mental state examination was notable for garrulous speech with a mildly impulsive and excited interruption on occasion, but no pressure of speech. No psychomotor changes were elicited. Affect was moderately bright, not expansive yet somewhat labile, not elevated, reactive and congruent. Thought was linear and logical; thought stream was preserved in rate, volume and consistency. Passive suicidality, but also future-oriented and hopeful themes, were apparent. No perceptual changes were elicited. Sensorium was notable for mild inattentiveness, difficulties with sequencing and inhibitory control, and planning, as described above. Her insight was excellent; judgement was acutely reasonable, albeit chronically impaired by her gamut of mild executive cognitive impairments.
Neuroimaging was undertaken. Her MRI brain revealed a left posterior fossa resection cavity and residual left cerebellar encephalomalacia consistent with her prior surgical history, mild periventricular chronic microvascular changes and scattered predominantly frontal cortical microvascular changes (Figure 1). There was no evidence of any thalamic or hypothalamic lesions, nor of any interval changes from her last MRI brain imaging 5 years prior. Magnetic resonance imaging (MRI) of Ms AC's brain in (A) axial, (B) coronal and (C) sagittal views, demonstrating the left posterior cerebellar resection cavity and encephalomalacia. Respectively, red arrows demonstrate involvement of: (A) the superior hemisphere of the left posterior cerebellar lobe; (B) the superior and inferior hemispheres of the left posterior cerebellar lobe; and (C) the posterior lobe of the left cerebellar vermis, and the lateral aspect of the left posterior cerebellar lobe, respectively from left to right.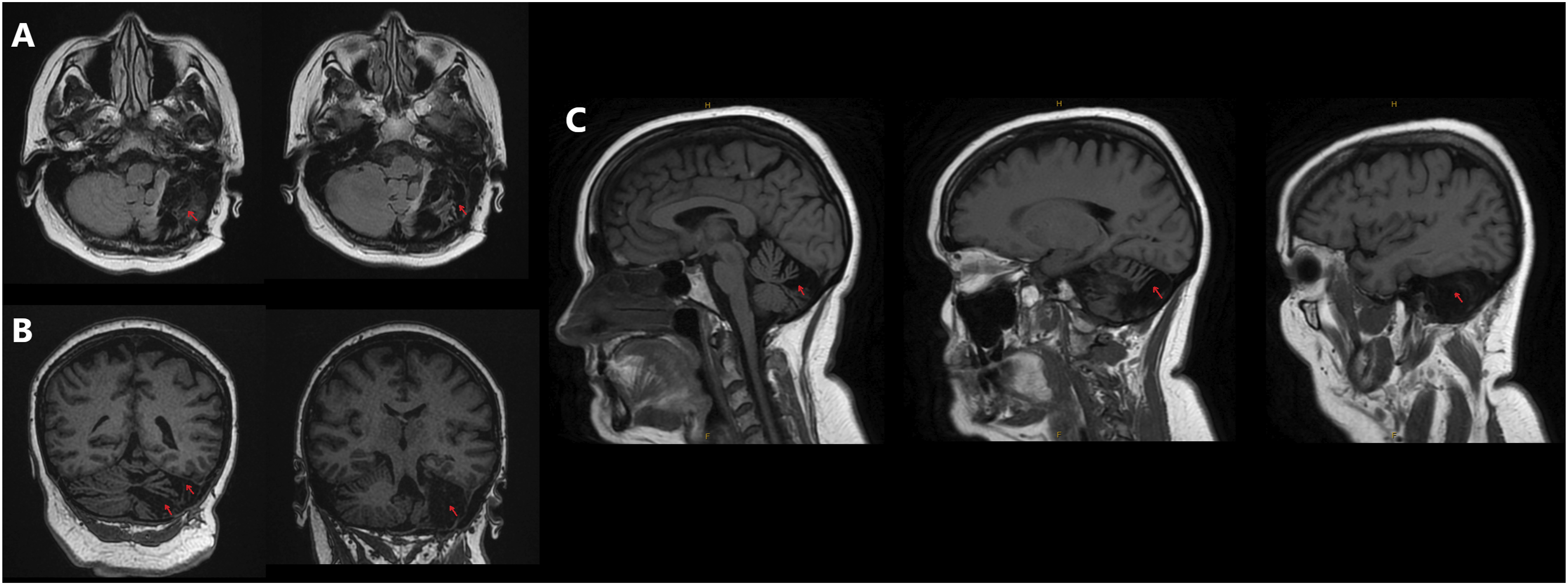
Further investigation was limited by Ms AC’s residence in an outer regional location. Local access to neuropsychology assessments, functional imaging and sleep studies, including polysomnography (PSG) and multiple sleep latency testing (MSLT), was not available. Ms AC declined baseline functional imaging and neuropsychological testing due to the associated travel time to a metropolitan centre and financial burden. PSG and MSLT were available in a nearer major regional centre; however, due to resource constraints, it was not feasible to arrange these studies contemporaneously with her sporadic episodes of hypersomnolence. Ms AC also declined urgent metropolitan referral for sleep studies during symptomatic periods, again due to the substantial travel and cost implications.
Collateral history from AC’s family members and her general practitioner corroborated the longstanding presence of AC’s difficulties with affective regulation, mild impulsivity, and static cognitive impairment, all without significant functional impairment. These informant histories similarly provided accounts of approximately week-long episodes of hypersomnolence occurring together with hyperphagia and disinhibited sexualised remarks, consistent with AC’s account. She had never experienced any issues with driving. She had undergone a lumbar puncture, including a neuronal autoimmune panel, which was unremarkable.
We reformulated AC’s baseline symptoms that had previously been described as hypomanic as manifestations of executive cognitive changes associated with CCAS. This diagnosis was supported by her scoring 6/10 on the Schmahmann CCAS scale, with scores above 3/10 considered diagnostic of CCAS. 3
AC’s cognitive impairments before and after commencement of lithium.
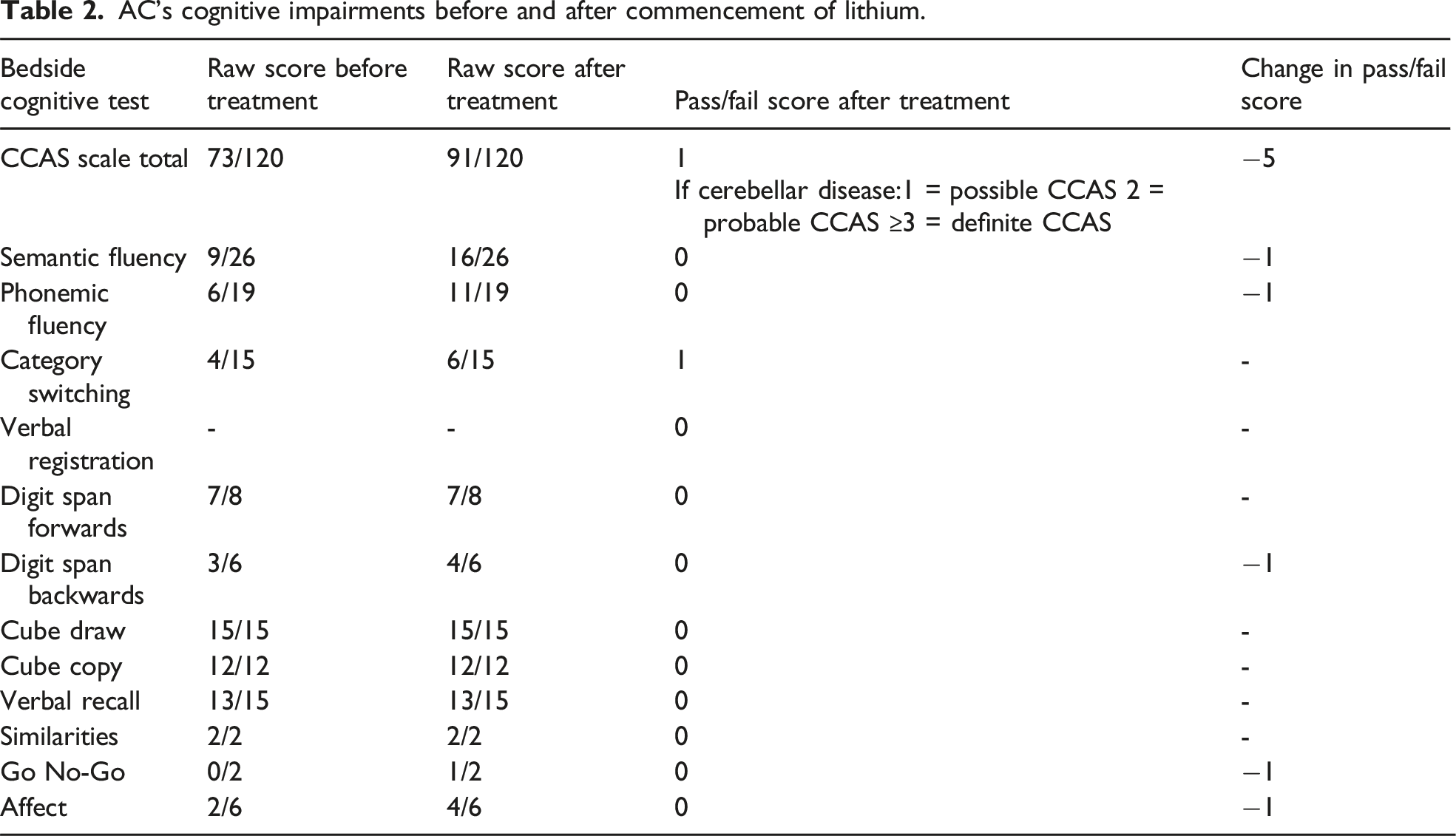
Interestingly, a delayed phase sleep pattern became more evident at this time, with AC frequently sleeping from 2-3am and waking at 10am. This sleep pattern responded well to the commencement of low-dose melatonin. Her olanzapine and venlafaxine were both gradually weaned without incident. No other pharmacological interventions were utilised, and while sleep hygiene was discussed with AC, she declined further behavioural or psychological interventions beyond supportive measures utilised on routine follow-up by members of her community mental health team. Her mental state has remained stable over the succeeding year.
Discussion
This report underscores the dilemma of complex diagnostication of coexisting affective and cognitive symptoms, and the inherent risk of employing broad strokes of Occam’s razor to an amorphous cluster of neuropsychiatric symptoms.
Both CCAS and KLS are rare disorders that are diagnosed on clinical criteria. While they are clinically well-characterized, they have yet to achieve mainstream recognition as distinct clinical entities and are frequently misdiagnosed.4-6
To our knowledge, this is the first description of both KLS and CCAS occurring in tandem; both likely precipitated by resection of an acoustic neuroma. This case underscores the merit of remaining diagnostically curious and reformulating a case as further information arises with longitudinal care, particularly as psychiatric diagnostic criteria and our understanding of neuropsychiatric illnesses continue to evolve.7,8 At the time of the BPAD diagnosis in this case, KLS remained a rare and newly characterized neurological illness, and CCAS had not yet been conceptualized by Schmahmann and colleagues. 9
Literature Review
Search terms used in our systematic review.

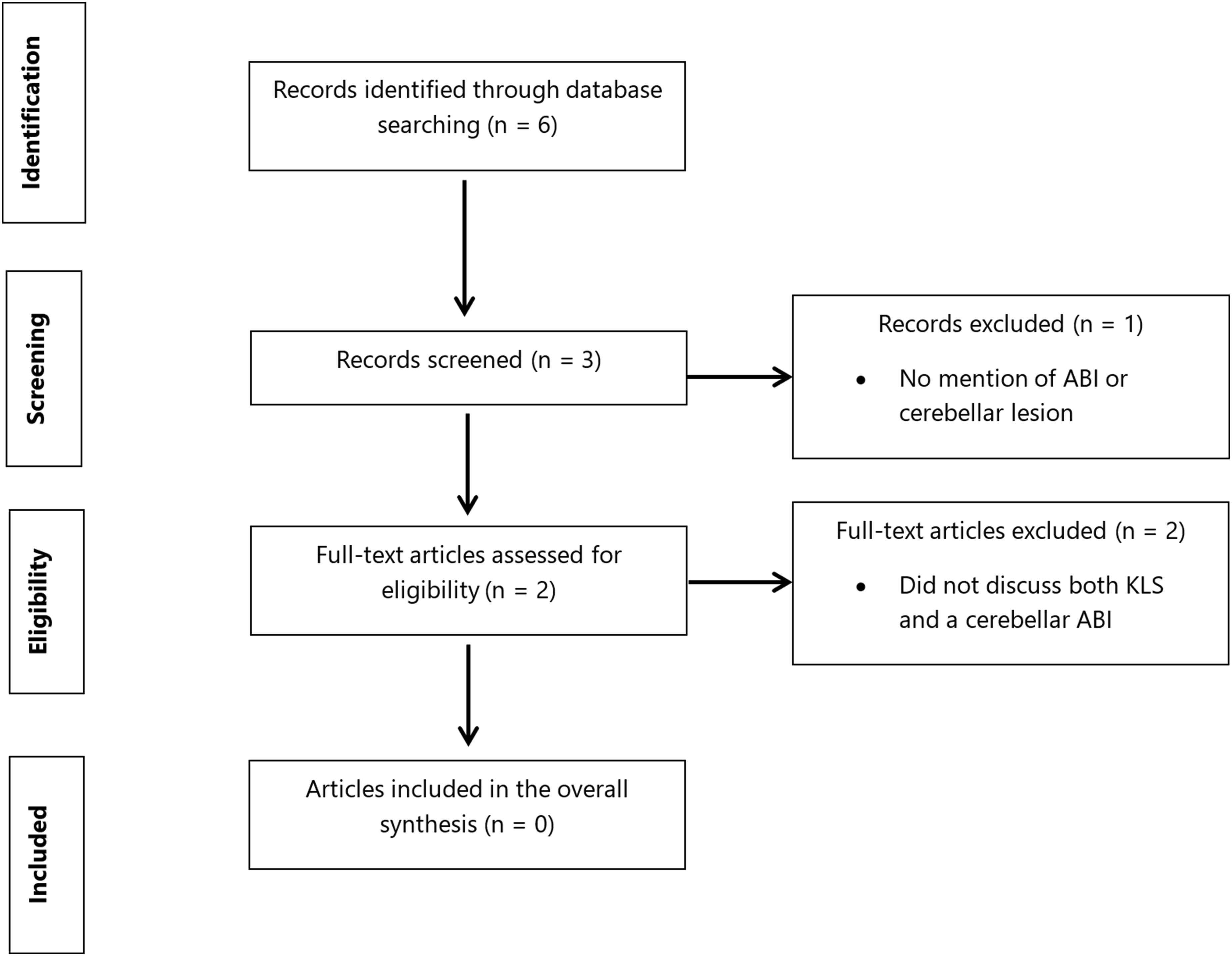
PRISMA systematic review flow diagram. The search strategy used Medical Subject Headings (MeSH) and free text terminology with Boolean operators, captured in Table 3, on the PubMed, MEDLINE, Embase, PsycINFO, and Cochrane databases from inception, together with handsearching of abstracts. Key inclusion criteria were that the article described a case of co-occurring Klein-Levin syndrome and cerebellar damage. Abbreviations: ABI, acquired brain injury; KLS, Klein-Levin syndrome.
Although not meeting inclusion criteria, we identified 1 case report describing KLS in association with mega cisterna magna, an anatomical variant affecting the posterior fossa. 10 In addition, we identified a functional connectivity study suggesting reduced connectivity between the cerebellum and thalamus during acute KLS episodes. 11 While these findings were not eligible for formal inclusion, they are noted here for context given their potential relevance to underlying mechanisms.
ABI is widely described as 1 possible precipitant for KLS in the literature, and was noted to be contemporaneously associated with 9% of cases in 1 study of 108 patients with KLS. 12 There is scant evidence or hypotheses in the KLS literature of potential underlying mechanisms for this. As AC demonstrated a specific form of ABI and contemporaneously developed symptoms consistent with KLS thereafter, we believe her unique case may prove helpful in illuminating areas for scientific interrogation and future research in KLS. Therefore, we have undertaken a comprehensive narrative review of KLS, CCAS, and their possible intersection. Our reference list is not exhaustive and prioritises original, contemporaneous papers of relevance to the broad scope and aims of this review. Furthermore, our hypotheses of potential mechanisms by which CCAS likely precipitated KLS in AC are speculative and based on the intersection of literature bases and theoretical underpinnings for both conditions.
Kleine-Levin Syndrome (KLS)
Natural History
Kleine-Levin syndrome (KLS) is a relapsing-remitting neuropsychiatric disorder that is often self-limiting over a decade. 4 However in 15%, the symptoms may continue indefinitely. The natural history of the syndrome is punctuated by recurrent episodes that typically last several days to weeks before spontaneous resolution, followed by full recovery for weeks to months in between episodes. KLS is characterized by abrupt-onset, episodic hypersomnia (>18 hours of sleep daily), compulsive hyperphagia, and variable, indiscriminate hypersexuality, irritability, dysphoria, derealization, apathy, regressed behaviour, disorientation, memory impairment, flu-like symptoms, and psychotic symptoms such as hallucinations and delusions.13,14 Hypersomnia, hyperphagia and hypersexuality constitute its classic triad.14,15 The diagnosis is fulfilled by clinical observation, or a dependable history of hypersomnia occurring with 1 other symptom of disinhibited behavioural, eating or cognitive disturbance, occurring over at least 2 distinct self-limiting episodes, in the absence of a better explanatory illness.16,17 It is a clinical diagnosis based on both reliable history and collateral information from family or carers, and does not require objective confirmation.17,18
In most cases, acute episodes are characterized not only by psychomotor slowing but also by a combination of attentional deficits and retrograde amnestic cognitive changes. Furthermore, on recovery, these individuals also often experience partial to complete retrograde amnesia for events occurring during these episodes. 19 Interestingly, cognitive and affective symptoms are thought to persist in an attenuated manner between episodes in up to a quarter of those with KLS.19-21
Risk Factors
KLS predominantly affects males in their second decade of life; however, it can occur at any age following a significant brain insult.13,22 The onset of KLS symptoms usually follows a febrile episode or a viral illness or gastroenteritis.5,13,23 Rarely, neurodevelopmental disorders, perinatal insults, and genetic susceptibilities have been reported to increase the risk of developing KLS.5,12,24-26 In 38-72 % of cases, infections have been identified as the precipitating factor for KLS.12,26 In the remaining cases, possible triggers include head injuries, stroke, other brain lesions, alcohol consumption, anaesthetic exposure, substance ingestion and menstruation.4,13,27 While evidence is limited, 1 hypothesis in the literature is that these triggers share a propensity to increase catabolism, open the blood brain barrier and promote local neuroinflammatory changes that may drive local accumulation of toxic byproducts or an alteration in affected neuronal tissue, particularly where an underlying genetic vulnerability to KLS exists. 25
Treatments
Lithium is widely cited as an effective treatment for KLS, with reported efficacies in treating acute episodes and preventing recurrent episodes.17,20,28–31 There is also some evidence for using other mood stabilizers, such as valproate and oxcarbazepine, as well as amantadine, clarithromycin, and IV methylprednisolone to terminate acute episodes.27,29,32 Stimulants such as methylphenidate and wakefulness-promoting agents such as modafinil are off-label, symptomatic treatments for excessive sleepiness, albeit of seeming limited efficacy in KLS and potentiating affective side effects such as irritability. 29 Psychosocial approaches to treatment include cognitive remediation, adaptations to work or school schedules, sleep hygiene psychoeducation, and avoidance of potential triggers such as sleep deprivation and alcohol.17,29
Differential Diagnoses and Mimics
Episodes of KLS often mimic encephalopathies, yet electroencephalographic, neuroimaging and laboratory findings typically do not demonstrate changes consistent with an encephalopathic state. 25 While transient limbic encephalitis secondary to autoimmune or paraneoplastic causes is frequently speculated to be a key cause of KLS in the literature – particularly given that viral illnesses frequently appear to precipitate episodes – the evidence to support a direct association has been circumstantial at best.20,24 Multiple sleep latency testing and electroencephalogram studies during acute episodes are frequently unremarkable and may show an increase in moderately fragmented, yet otherwise relatively normal sleep.17,24,31,33 Reduced slow wave sleep was found in the first half of acute episodes in 1 study, but did not correlate to symptom onset or duration. 34 It has been hypothesised to be a disorder of sleep satiety. 24
Both BPAD and KLS exhibit a relapsing-remitting pattern of illness, with common triggers, shared circadian rhythm and neurogenetic vulnerabilities, and responsiveness to mood stabilisers, suggesting shared underlying pathophysiological mechanisms. Additionally, they both involve deficits in attention, retrieval, and verbal memory. However, KLS lacks hallmark manic and depressive symptoms on mental state examination that characterise BPAD. 2
Key differentials for KLS-like presentations include psychiatric disorders such as BPAD (including cyclothymia and mixed state presentations) and atypical depression; and neurological disorders such as narcolepsy, hypothalamic and third ventricular lesions from eg, stroke or tumours, non-convulsive status epilepticus, basilar migraines, atypical presentations of idiopathic hypersomnia with variable sleep times, Kluver-Bucy syndrome, metabolic disorders such as intermittent porphyria and ornityl-carnitine transferase deficit, and autoimmune encephalitis, eg, anti-IgLON5 disease.12,17,35,36 Some cases are obfuscated by the comorbidity of both KLS and a typical mimic syndrome, such as BPAD.21,32 KLS is distinguishable from its mimics and differential diagnoses by an abrupt onset of severe hypersomnolence (eg, >18 hours of sleep per day) that is ultimately self-limiting and has episodic recurrence. 37 Careful dissection of the symptomatology of these presentations is essential to timely, accurate diagnostication.
Cerebellar Cognitive Affective Syndrome (CCAS)
Natural History
Cerebellar cognitive affective syndrome (CCAS), also known by its eponymous name ‘Schmahmann’s syndrome’, is similarly a syndrome characterized by affective and cognitive changes. It was first characterized in 1998 9 as damage to the posterior cerebellum with consequent impairment in executive, visuospatial, and linguistic function, and affective regulation.3,6,38-40 It has been conceptualized as a ‘dysmetria of thought’.38,41 Unlike KLS, where working and declarative memory are impaired, but executive functions are preserved, 42 the most affected cognitive domain in CCAS is executive function itself. 19 It can occur as a consequence of posterior cerebellar injury, with no predominant cause identifiable in the literature to date. Common causes include stroke, tumours, disruption to cerebellar-cortical pathways or cerebellar integrity from common neurodegenerative illnesses such as Alzheimer’s disease and frontotemporal dementia, congenital cerebellar malformations and neurogenetic conditions such as Joubert syndrome and Dandy-Walker malformation, and the direct impact of rarer dementias such as the spinocerebellar ataxias.3,43-46
Differential Diagnosis and Mimics
The affective instability that accompanies CCAS can be readily mistaken for that associated with borderline personality disorder. 47 It similarly mimics other neuropsychiatric conditions with impulsivity or lability as key symptoms, including attention deficit-hyperactivity disorder (ADHD), other forms of BPAD such as cyclothymia and bipolar affective disorder type II, acquired brain injuries (including those not affecting the cerebellum), impulse control spectrum disorders, and pseudobulbar affect accompanying other neuropsychiatric disorders such as multiple sclerosis, stroke, and amyotrophic lateral sclerosis.48-51
The dysexecutive cognitive impairments associated with CCAS, particularly superimposed on its affective instability, warrant scrutiny to exclude illnesses that may present similarly. This includes BPAD; 30-60% of those with this disorder have persistent cognitive deficits that are similar to yet typically less severe than the broad cognitive impairments common in schizophrenia, including in verbal learning, processing speed, explicit memory, and attention in addition to the executive dysfunction also evident in CCAS.52,53 It is also prudent to exclude neurodegenerative illnesses such as subcortical vascular dementia and behavioural variant frontotemporal dementia (bvFTD), and its mimics, such as the so-called ‘phenocopy’ bvFTD syndrome that may represent a ‘pseudodementia’ of reversible primary psychiatric illness.54,55
Treatments
Lithium can be used in both KLS and CCAS. While lithium has been shown to abort acute episodes and prevent recurrence in KLS, its role in CCAS is more symptomatic, where it may be prescribed for affective instability, impulsivity and suicidal ideation.47,56-60 Lithium has a notable high tropism for the cerebellar cortex, as evidenced by the well-characterised acute and chronic anterior cerebellar toxidromes.61-63
The body of evidence for effective treatments in CCAS is limited. There are case reports of symptomatic improvement with low-dose aripiprazole, modafinil, low-dose zolpidem, dextromethorphan/quinidine, donepezil, memantine, low-dose quetiapine, and a combination of lithium, clozapine and clomipramine introduced in tandem.47,64-70
Case Discussion
In this case, AC demonstrated mild-to-moderate and circumscribed executive impairments; her excellent abstraction, for example, demonstrated some of her executive functions remained intact. Some circumscribed linguistic abilities were also mildly impaired, consistent with the literature on language and visuospatial cognitive changes commonly accompanying CCAS. 3 Her personality changes and affective regulation difficulties following her acoustic neuroma excision were both notable but not unexpected, given the significant left cerebellar resection cavity and encephalomalacia observed on neuroimaging, including involvement the cerebellar vermis. However, once BPAD was diagnosed – prior to the recognition of KLS or CCAS as clinical syndromes – this diagnosis remained with AC.
For decades, significant diagnostic overshadowing persisted, with her personality changes, affective symptoms, and impulsive features of her executive cognitive impairments being characterized as an indolent, pervasive hypomania. Her episodes of hypersomnolence, hyperphagia and hypersexuality were attributed to depressive or mixed states. Her cognitive and affective symptoms, and periodic episodes of hypersomnolence, never reached severity thresholds requiring hospitalisation, and so her diagnosis remained unchallenged for many years. When she first presented to the public mental health services, her suicide attempt, chronic impulsivity and mild affective lability was diagnosed as borderline personality disorder- a reflection of modern diagnostic heuristics that often leads to diagnostic overshadowing.48,71
Her response to low-dose lithium supports the view in the literature of its efficacy in KLS, and in dysexecutive impulsivity as occurs in CCAS.70,72 A low dose would not result in rapid antimanic or antidepressant effects, 73 as would be expected if she truly had BPAD.
AC’s development of CCAS with her acoustic neuroma resection temporally coincides with her development of KLS. This case provides a practical and illustrative example of the impact of injury to brain regions in broader neuronal connectivity networks on the functioning of otherwise undamaged cerebral areas: for example, how damage to the posterior cerebellum may have disrupted thalamic functioning in AC’s presentation. Furthermore, AC’s left-sided cerebellar lesion may have potentially impacted the contralateral, right cerebral hemisphere, crucial for social cognition and emotional regulation relative to the left, 74 thereby predisposing her to developing mild lability and a more extraverted, less socially inhibited personality change.
The thalamus and hypothalamus are implicated in the pathogenesis of KLS in at least some cases.4,19,24,75 A functional connectivity study demonstrated impaired connectivity between thalamus and cerebellum, and thalamus and dorsal pons, during an acute episode of KLS. 11 Functional neuroimaging further suggests that executive and associative networks, including the thalamus and to lesser extents the hypothalamus, caudate nucleus, orbitofrontal cortex, anterior cingulate cortex, and superior and medial (including hippocampal) temporal regions, are impaired between acute episodes of KLS. 17 However, there appears to be compensatory overactivity in parts of these networks during task performance, which helps to limit executive cognitive impairment, but may result in reduced attention and processing speed. 19 KLS also has notable overlap of neurogenetic risk factors with circadian disorders and BPAD, 76 which is notable given AC’s delayed phase sleep.
Similarly, the cerebellum is now understood to have important roles not only in coordination, but also cognition and affect via upstream connectivity with the neocortex and limbic system respectively through its thalamic projections (Figure 3).77-81 While cerebellar lesions often clinically manifest as ataxia, lesions to the lateral hemisphere of the posterior (‘associative’) cerebellum can also lead to cognitive impairment, and those to the posterior cerebellar vermis – the so-called ‘limbic cerebellum’ – may further induce affective changes.38,82 As a modulator of brain activity, damage to these posterior cerebellar structures can result in an ‘dysmetria’ of affect and thinking, as AC experienced. Additionally, the cerebellum connects with and is thought to regulate the DMN.83,84 Erosion of cerebellar connectivity with the DMN impacts its cognitive modulatory abilities, especially executive functions.
84
Furthermore, disrupted DMN connectivity is also implicated in idiopathic hypersomnia.
85
The posterior cerebellum – implicated in cerebellar cognitive affective syndrome (CCAS) – mediates affect and cognition through its projections to the thalamus and subsequently, thalamic connections to the limbic system and neocortex. Thalamic dysfunction is implicated in at least some cases of Kleine-Levin Syndrome (KLS).4,19 Neural network disruption through damage to the posterior cerebellum may precipitate KLS, in addition to CCAS. Cerebello-thalamo-cerebro-cortical circuits represent an integrated network which include the cerebellum (posterior-inferior cerebellum, PIC) via the deep cerebellar nuclei of the superior cerebellar peduncle (SCP), the contralateral red nucleus (RN), and the anterior thalamic nucleus (AnT), which then goes on to connect to multiple cortical areas. Limbic regions communicate with the cerebellum via the anterior pontine nucleus (APN). AC’s lesion extensively involves the PIC which together with the AnT, is highlighted in red. Adapted from D’Angelo and Casali, 2012. Created in BioRender. https://BioRender.com/7xp5tov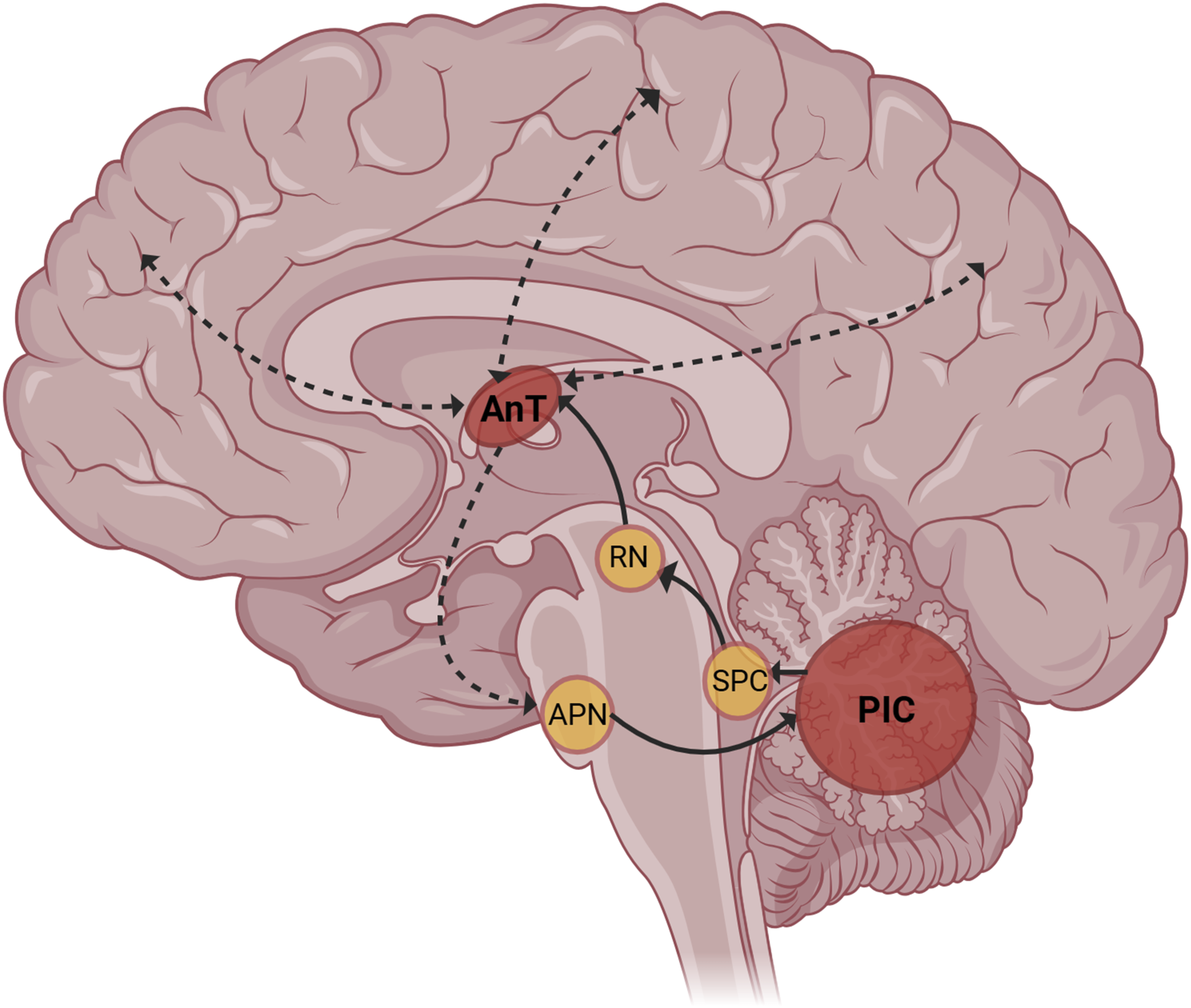
Therefore, it is possible that disrupted cerebellar-thalamic and cerebellar-DMN connectivity, occurring in the setting of significant left cerebellar damage that diagnostically constellated as CCAS, may have posed key risk factors for AC to develop KLS.
Our review of the literature has several key limitations. There is a paucity of mechanistic evidence describing how ABIs may cause KLS, limited data on the symptomatic treatment of CCAS, and minimal evidence beyond case reports or small case series regarding treatments for KLS. Furthermore, our systematic review did not identify any studies examining the intersection of these conditions. In light of these gaps, we adopted a narrative review with broad inclusion criteria to synthesis the available literature. This does not constitute an exhaustive overview of either KLS or CCAS in isolation, incorporates evidence from heterogeneous sources and disciplines and carries a risk of interpretation bias.
Similar limitations apply to the diagnostic process. The diagnosis of probable KLS – a clinical diagnosis of exclusion – was based on retrospective record review and informant history, which may introduce recall and interpretation biases. KLS remains a diagnosis of exclusion; while circadian rhythm disorders typically do not present with the same magnitude of unprovoked, episodic, time-limited changes in sleep, nor the associated features of hyperphagia and hypersexuality, our inability to definitively exclude alternative sleep disorders given our restricted access to sleep studies in this case is a notable limitation. Likewise, the diagnosis of CCAS remains a clinical diagnosis and is subject to similar bias, although a structured bedside cognitive assessment provided further support for this diagnosis.
ABIs are 1 known cause of KLS. 13 In this case, the incidence of CCAS, a specific ABI syndrome with characterised neuroconnectome correlates, offered a unique opportunity to hypothesise some possible mechanisms of developing KLS based on contemporary understanding of both conditions. Research that characterises the nature of ABIs that have temporally coincided with the onset of KLS, including further functional connectivity studies and tractography, would be helpful in interrogating our hypothesis.
Conclusion
AC’s development of CCAS with her acoustic neuroma resection temporally coincides with her development of KLS. This case provides an illustrative example of the impact of injury to brain regions in broader neuronal connectivity networks on the functioning of otherwise undamaged cerebral areas: particularly how damage to the posterior cerebellum may have disrupted thalamic and DMN functioning in AC’s presentation.
This case highlights the complex, domino effect central nervous system lesions can have on the connectome, and the risk of being inadvertently reductionistic toward neuropsychiatric syndromes of mixed cognitive and affective symptoms when a patient has a history of mental illness. We hope this report will help inspire clinical curiosity, appetite for complex and ongoing formulation, and research into the intersection of these complex conditions, which could pose benefit for disorders with shared risk factors like BPAD.
Footnotes
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.