
Abstract
High bone-localized concentrations of antimicrobial agents are necessary for the long-term effective treatment of chronic osteomyelitis, particularly in cases of severe infection and bone loss. This study addressed infection control and bone regeneration simultaneously using hydroxyapatite and natural biopolymers. Moxifloxacin hydrochloride was delivered via composite scaffolds produced from polyvinyl alcohol/gelatin and hydroxyapatite with potential applications in osteomyelitis treatment and bone tissue engineering. The composite scaffolds exhibited a well-defined porous architecture, characterised by macropores (≥100 µm) and micropores (≤20 µm), facilitating cellular infiltration and drug loading. Biomineralization and cell culture assays were used to evaluate the scaffold’s bioactivity and biocompatibility. Analyses of mineralized scaffolds using Fourier-transform infrared spectroscopy and scanning electron microscopy revealed HA nucleation on the scaffold’s surface after immersion in simulated bodily fluid for varied time points. Protein adsorption and haemolysis tests were conducted to confirm the blood compatibility of scaffolds. Cell culture studies using human mesenchymal stem cells indicated non-cytotoxicity and robust cell adhesion. These findings suggest the potential suitability of these scaffolds for future clinical applications in the treatment of chronic osteomyelitis and bone regeneration.
Introduction
Staphylococcus aureus (S. aureus) infections are the main cause of osteomyelitis (OM), a severe, progressive inflammatory disease. 1 Conventional treatments for OM include surgical debridement of necrotic bone tissues, antiseptic irrigation, and systemic antibiotic administration for roughly 4–6 weeks. 2 However, there are various drawbacks associated with this method. For example, extensive debridement may create voids in bone structures, requiring grafting and posing the risk of further infection. 3 Systemic side effects, pain, discomfort, and high costs are associated with long-term intravenous use of antibiotics. Additionally, only a fraction of the administered antibiotics reaches the bone. It is reported that conventional therapeutic approaches are effective in only 50%–70% of chronic OM cases. 4 Drug-loaded scaffolds, which combine local intra-osseous antimicrobial delivery with regeneration assistance, offer a viable solution to these problems. Scaffolds produced from various materials, including biopolymers, bioactive glasses, and ceramics, loaded with antibiotics such as vancomycin, gentamicin, or tobramycin, have been explored for this purpose. 5 By providing prolonged release at the infection site, these scaffolds increase local medication concentrations while reducing systemic side effects. However, the inherent brittleness of contemporary bone scaffolds, especially those composed of ceramics like hydroxyapatite (HA), β-tricalcium phosphate (β-TCP), and bioactive glass, limits their applications. 6 Long-term bone regeneration is further hampered by the mechanical mismatch between these brittle materials and bone’s inherent elastic nature, as the scaffolds are unable to withstand physiological stresses during the healing process.
Non-toxic and biodegradable polymers, e.g., polyvinyl alcohol (PVA) and gelatin, are known for their applications as scaffolds in bone tissue engineering, drug transporters, and implant materials.7–9 PVA reduces the brittleness that comes with ceramics and improves the mechanical qualities of scaffolds by making them more flexible and durable. In addition to providing structural support, combining these polymers with ceramics allows for customization of the porosity required for the bone constructs while concurrently serving as an effective platform for drug delivery. Their exceptional bioactivity, effective interaction with biological entities, osteoconductivity, osteoinductivity, and non-toxicity render them promising materials for the fabrication of implantable scaffolds in bone tissue engineering.10,11
To date, several fabrication techniques, such as gel casting, freeze casting, foaming, and salt leaching,12,13 have been established to incorporate macroporosity within HA/polymer-based scaffolds.14,15 Microporosity (≤20 µm) and macroporosity (≥100 µm) are both essential in bone tissue engineering. Macroporosity support cell migration, vascularization, and tissue ingrowth, while microporosity promotes protein adsorption, cell attachment, and nutrient diffusion, together creating the perfect environment for bone integration and regeneration.16,17 This dual-porosity approach improves infection management and bone regeneration by allowing vascularization through the large pores and regulated medication release from the small pores. Larger pores can accommodate large drug quantity and enable a quicker initial release, which is advantageous for obtaining high local antibiotic concentrations immediately after implantation to combat acute infection. While the smaller pores offer a large surface area for antibiotic adsorption, leading to higher drug-loading capacity. This approach highlights the value of multi-scale porosity in bone scaffold design.
This study aimed to fabricate HA-PVA-Gelatin scaffolds with multi-scale porosity using a foaming and freeze-drying technique. The scaffolds were analysed for controlled drug release characteristics. To produce medicated composite scaffolds with multiscale porosity, moxifloxacin hydrochloride (MOX), a fourth-generation fluoroquinolone antibiotic that is frequently used in the treatment of OM, was loaded into the scaffold. Physical characteristics, drug release profile, blood compatibility, cell survival, and bioactivity of the scaffolds were examined in detail. The overarching objective was to evaluate scaffolds potential for future therapeutic applications within the domains of bone regeneration and OM therapy.
Experimental details
Materials
Analytical-grade calcium nitrate (Ca(NO3)2.4H2O) and diammonium hydrogen phosphate ((NH4)2HPO4) (AppliChem, Germany) were used as precursors for HA synthesis. Gelatin (BDH, UK), Tween 20 (Merck, Germany) and PVA (Merck, Germany, MW: 72,000) were used in the fabrication of scaffolds. Lysozyme (Carbo synth) with an enzymatic activity of 10,000 U/mL was used for the biodegradation study. Moxifloxacin (MOX, Merck, Germany) was used as the model drug in the drug release experiments. Bovine Serum Albumin (BSA powder, CAS no 9048-46-8) was obtained from Merck for protein adsorption experiments. Phosphate-buffered saline (PBS; pH 7.4, gibco) was used as a simulated biological medium for swelling, degradation and drug release studies. The following reagent grade chemicals and distilled water were used for the preparation of SBF: Sodium chloride (NaCl, Fisher Scientific), sodium hydrogen carbonate (NaHCO3, Fisher Scientific), potassium chloride (KCl, Fisher Scientific), di-potassium hydrogen phosphate trihydrate (K2HPO4.3H2O, Merck), magnesium chloride hexahydrate (MgCl2.6H2O, Merck), calcium chloride (CaCl2, Merck), sodium sulfate (Na2SO4, Merck), Tris-hydroxymethyl aminomethane((HOCH2)3CNH2, Merck),1
HA synthesis
HA powder was synthesised using a microwave-assisted wet precipitation method. 18 Briefly, diammonium hydrogen phosphate ((NH4)2HPO4) (AppliChem) and calcium nitrate (Ca(NO3)2·4H2O) (UniChem) were used as precursors. All calculations were based on the Ca/P ratio of 1.67. Calcium nitrate (1 M) and diammonium hydrogen phosphate (0.6 M) solutions were made independently in 400 ml of distilled water. Prior to mixing, ammonium hydroxide (BDH) was added to both solutions to raise their pH above 10. (NH4)2HPO4 solution was added dropwise to the calcium nitrate solution at a rate of 2 ml/min. The reaction mixture was refluxed in a microwave oven (Samsung MW101P) for 1, 3, 5, and 10 min (30 s ON, 30 s OFF) at 600 W, 850 W, and 1000 W. The reaction mixture was then filtered, washed with distilled water, and HA precipitates were dried in an oven at 80°C for 2 h. After grinding, the obtained powder was then dried in a box furnace at 200°C for 2 h, heat treated at 1200°C for 2 h (ramp rate: 5°C/min) and cooled down to room temperature (ramp rate: 30°C/min) for further use.
Scaffolds fabrication
Compositions, porosity values, and mechanical properties of the fabricated scaffolds.
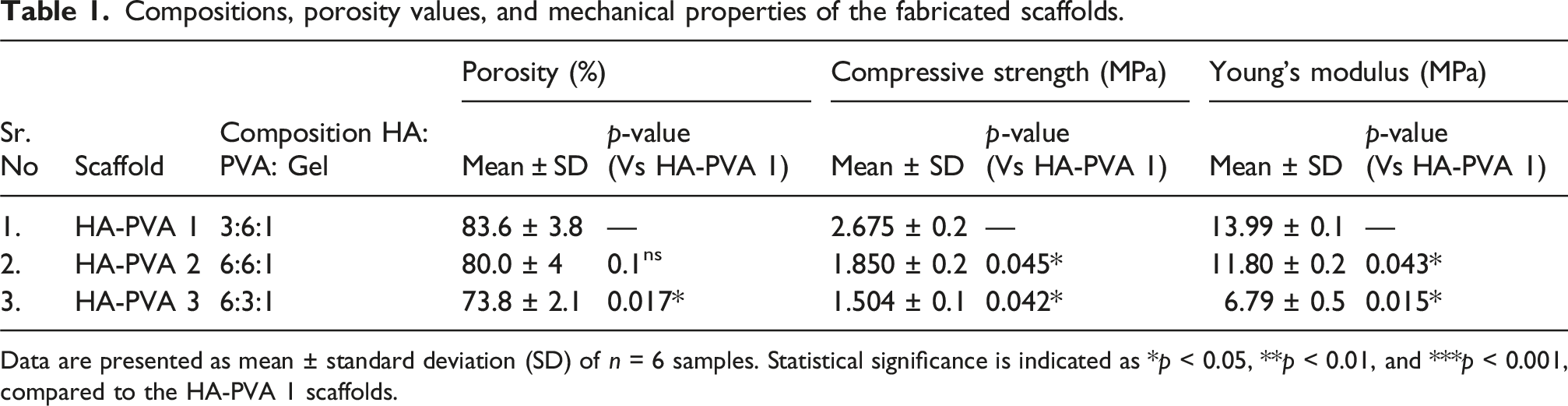
Data are presented as mean ± standard deviation (SD) of n = 6 samples. Statistical significance is indicated as *p < 0.05, **p < 0.01, and ***p < 0.001, compared to the HA-PVA 1 scaffolds.
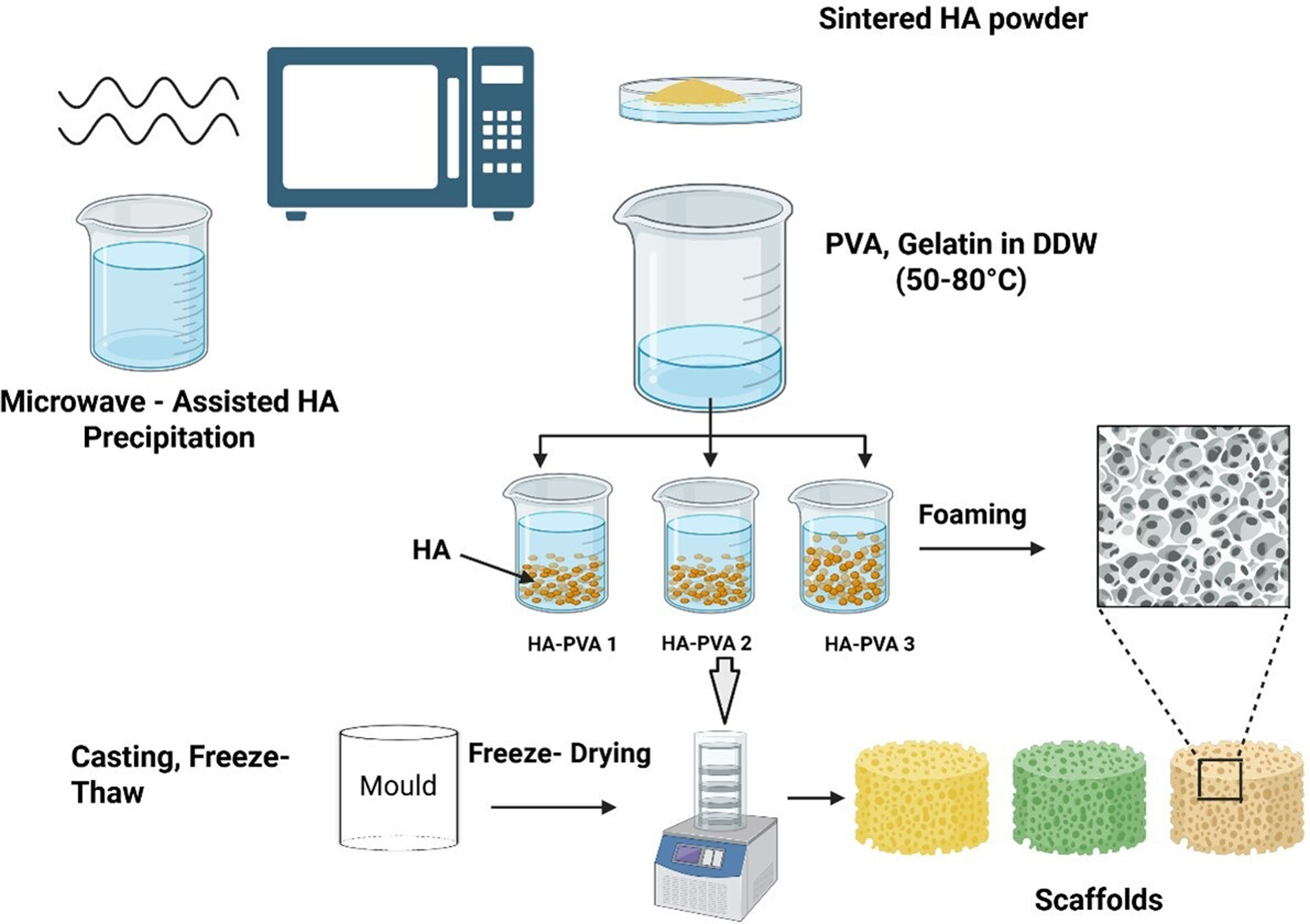
Schematic representation of the fabrication process for HA–PVA–Gelatin scaffolds.
For preparing HA-PVA 1 and HA-PVA 2, 6 g of PVA was dissolved in 100 mL of DDW at 80°C under continuous stirring (600 rpm) until a clear solution was obtained (∼30 min). For HA-PVA 3, 3 g of PVA was dissolved under identical conditions. After cooling the PVA solution to 50°C, 1 g of gelatin was added and dissolved completely in each formulation. Then heat treated HA powder (3 g for HA-PVA 1 and 6 g for HA-PVA 2 and HA-PVA 3) was gradually dispersed into the PVA–gelatin solution in small portions while stirring at 40°C (500 rpm) to ensure uniform distribution of HA particles. Tween 20 (50 µL) was then added as an emulsifier in each suspension and was mechanically stirred at 800 rpm for 15 min until stable foam formation. The resulting foam was then casted into cylindrical-shaped moulds (5 × 5 mm) to produce scaffolds and subjected to five consecutive freeze-thaw cycles: freezing at −20°C for 12 h, then thawing at 20°C for 12 h. Finally, to remove residual Tween 20, scaffolds were immersed in DDW for 7 days (daily water replacement) and subsequently freeze-dried (−40°C, 100 m Torr, 24 h, VirTis Benchtop lyophiliser; cooling rate: 1°C/min) to generate macropores within the scaffold structure.
Methods
FTIR
The FTIR was used to characterise the chemical composition of composite scaffolds and HA (n = 3). The technique is essential to verify the presence of certain functional groups associated with both HA and polymer components. Nicolet 6700, USA instrument was used in photoacoustic mode. Spectra were acquired over a wavenumber range of 4000 to 450 cm−1 with a resolution of 8 cm−1. Each spectrum was collected by averaging 256 scans to improve the signal-to-noise ratio.
XRD
The crystalline structure and phase composition (n = 3) of the pure HA and its composites were characterised using X-ray diffraction (XRD) analysis. The XRD measurements were performed on a PERT-PRO diffractometer system equipped with a Cu Kα radiation source (λ = 1.5406 Å). Before analysis, the scaffolds were pulverised to ensure homogeneity. Diffraction patterns were acquired over a 2θ range of 10° to 80° with a step size of 0.05°.
SEM
The structural morphology (n = 3) of the scaffolds was characterised using scanning electron microscopy (SEM, JEOL JSM6490A, Japan). SEM images were acquired at an accelerating voltage of 10–15 kV. Before examination, scaffold cross-sections were prepared using a razor blade and sputter-coated with gold to enhance conductivity. The pore sizes were quantified using ImageJ software (version 1.53, National Institutes of Health, USA).
Porosity and mechanical properties
The porosity of the scaffolds was determined using a liquid displacement method, with absolute ethanol serving as the displacement medium.
19
The percentage porosity was calculated for each scaffold composition using the following equation: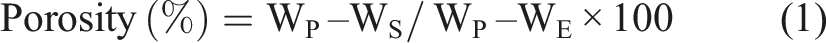
WS is the weight of the dry scaffold, WP is the weight of the scaffold with ethanol-filled pores, and WE is the weight of the scaffold suspended in ethanol. All tests were done in triplicate (n = 3). Compression testing (n = 6) was conducted using a universal mechanical tester (M 500-50AT) equipped with a 15 N load cell. Scaffolds were prepared as rectangular blocks measuring 12 × 6 × 6 mm3 (l × w × h). Unconfined compression tests were performed under ambient conditions at a crosshead speed of 1 mm/min until 20% strain was achieved. The compressive strength was defined as the maximum compressive stress, while the effective elastic modulus was calculated from the linear region of the stress-strain curve.
In-vitro tests on scaffolds
Scaffolds were tested for in-vitro biomineralization, degradation, protein adsorption, drug release and cell viability. Before each test, scaffolds were disinfected by immersion in 70% ethanol for 30 min. For cell culture and antibacterial testing, the samples were sterilized by immersion in 70% ethanol, followed by exposure to UV light for 1 h to ensure aseptic conditions.
Biomineralisation
The biomineralisation behaviour of the scaffolds was evaluated using the SBF immersion test. The SBF solution was prepared according to the method proposed by Kokubo. 20 5 × 4 mm2 rectangular samples (n = 3) of each scaffold composition were immersed in SBF and incubated at 37°C for up to 28 days. The SBF solution was refreshed every 3 days throughout the experiment. At predetermined time points (1, 7, 14, and 28 days), the scaffolds were removed from the SBF solution, gently washed with deionised water followed by 70% ethanol, and dried at 50°C for 2 h. The samples were then allowed to dry further at room temperature for 7 days. The formation of an apatite layer on the scaffold surfaces was assessed using SEM and FTIR.
Swelling behaviour
To investigate the swelling behaviour of scaffolds, samples were prepared by cutting them into uniform pieces (approx. 10 × 5 mm2, rectangular) with equal dry weights (Wd). These samples were immersed in PBS solution at pH 7.4 and maintained at 37°C. At predetermined intervals (Day 7, Day 14, and Day 21), the scaffolds were carefully removed from the solution, gently blotted using filter paper to eliminate excess surface water and weighed to determine their wet weights (Ww). The degree of swelling (DS) was calculated for triplicate samples (n = 3) using the following equation:
Degradation behaviour
The in-vitro degradation behaviour of the scaffolds (n = 3) was evaluated in PBS (pH 7.4) containing lysozyme at 37°C. Samples of pre-weighed scaffolds were immersed in PBS supplemented with lysozyme (10,000 U/mL) and incubated at 37°C for up to 21 days. The initial dry weight of each scaffold was recorded as Wi. At predetermined time points (days 7, 14, and 21), scaffolds were removed from the medium, freeze-dried, and weighed to obtain the final dry weight (Wt). The degradation rate was calculated using the following equation: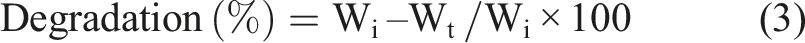
Protein adsorption
The protein adsorption experiments were conducted on scaffolds (n = 3) using a modified version of a previously reported method.
21
Bovine Serum Albumin (BSA) solution (4 mg/mL) was prepared in PBS. Each scaffold composition (0.3 g) was equilibrated with PBS for 24 h before the experiment. The pre-weighed, fully swollen scaffolds were immersed in the BSA solution and subjected to high vacuum conditions (−60 cmHg) at 37°C. Supernatants were collected at 2, 4, 8, and 24 h intervals for analysis. Protein quantification was performed using the Bradford assay. Briefly, 0.1 mL of the supernatant was mixed with 3 mL of Bradford reagent (Sigma-Aldrich, Product No. B6916). The BSA concentration was determined using UV-visible spectroscopy (Perkin Elmer Lambda 25, Cambridge Scientific, UK) at 595 nm, using a pre-established standard curve. The amount of adsorbed protein was calculated using the following equation: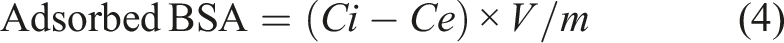
Haemolysis
Blood haemolysis tests were conducted on prepared scaffolds (n = 3) with equal weights, following previously established protocols.
22
Human blood samples, collected in EDTA-containing tubes, were obtained from healthy volunteers after receiving approval from the local ethics committee of COMSATS University Islamabad. Positive and negative controls were prepared by adding 0.25 mL of blood to 2 mL of distilled water and 0.9% saline solution, respectively. The optical density (OD) of the samples was measured at 545 nm using a Lambda 25 UV/Vis spectrophotometer (Perkin Elmer, UK). The percentage of haemolysis was calculated using the following equation: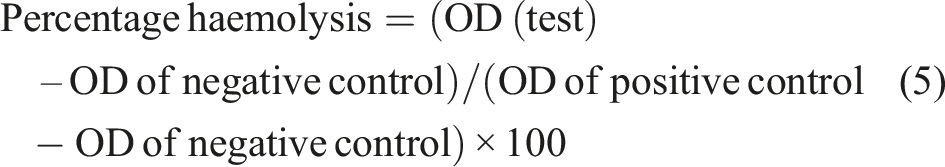
Moxifloxacin release and antibacterial activity
Scaffolds of each composition were sectioned into 1 g pieces (8 × 4 × 2 mm) and loaded with MOX by immersion in 5 mL of a 1 mg/ml MOX solution in PBS for 48 h. The drug-loaded scaffolds were then retrieved, and the amount of MOX incorporated was determined by calculating the difference in drug concentration before and after immersion. Drug release studies were conducted by immersing the MOX-loaded scaffolds (n = 3) in 5 mL PBS at 37°C under gentle agitation (60 rpm) for 72 h. Aliquots were collected at predetermined time points (1, 6, 24, 48, and 72 h). The concentration of released MOX was quantified using a UV-visible spectrometer (Perkin Elmer Lambda 25, UK) at 289 nm. 23 Before analysis, the stock solution of MOX and supernatant solutions from each scaffold were diluted with PBS at a 1:50 volume ratio. A standard curve was generated using known concentrations of MOX (1–18 µM) in PBS to determine the percentage of drug released.
The antibacterial activity (n = 3) of the drug-loaded scaffolds was evaluated using S. aureus (gram-positive) as a model organism. The zone of inhibition (ZOI) was measured to assess the antimicrobial efficacy. Oxoid Moxifloxacin antimicrobial Susceptibility discs (Thermo Fisher Scientific, UK) were used as positive controls. Blank scaffold without drug loading was used as negative control. Detailed experimental conditions and methods for the ZOI assay are provided in the Supporting Information (SI).
Cell viability
The cell culture experiments (n = 3) used human mesenchymal stem cells (hMSCs) obtained from Merck, Germany. Before cell seeding, scaffolds underwent a sterilisation procedure involving immersion in 70% ethanol followed by exposure to UV light for 1 h. Subsequently, 30,000 cells were seeded onto each scaffold in a 12-well plate. Cell viability and proliferation were assessed using the Alamar Blue Cell Viability Assay (Fisher Scientific, UK), on days 1, 4, and 7 of the experimental timelines. The Alamar Blue reagent was added to the cell culture medium, and the plates were incubated at 37°C for 1–4 h. Absorbance measurements were then acquired using a microplate reader at 570 nm. As a control to compare cell adhesion and viability, hMSCs were also seeded on standard tissue culture plates (TCP). All experiments were performed in triplicate.
Statistical analysis
The experimental data from all studies were analysed using Analysis of Variance (ANOVA). A p-value of ≤ 0.05 was considered statistically significant. Data are reported as mean ± standard deviation for n = 3 samples.
Results and discussion
FTIR
Figure 1(a) displays the FTIR spectrum of HA powder sintered at 1000°C. The spectrum exhibits characteristic peaks associated with HA,
18
including the hydroxyl (OH) stretching vibration at 3571 cm−1 and the phosphate (PO4−3) group vibrations.24,25 Specifically, the PO4−3 bending modes are observed at 571–601 cm−1, while the stretching modes appear at 960–1092 cm−1. The presence of these distinctive HA peaks, coupled with the absence of extraneous signals, confirmed the purity of the produced HA powder. The FTIR spectra presented in Figure 1(b) comprising pure PVA and gelatin used in scaffolds synthesis. (a) FTIR spectrum of sintered HA, (b) FTIR spectra of PVA and gelatin, (c) FTIR spectrum of freeze-dried HA-PVA-Gel scaffolds, (d) XRD of pure HA powder, and (e) XRD of porous freeze-dried HA-PVAGel scaffolds.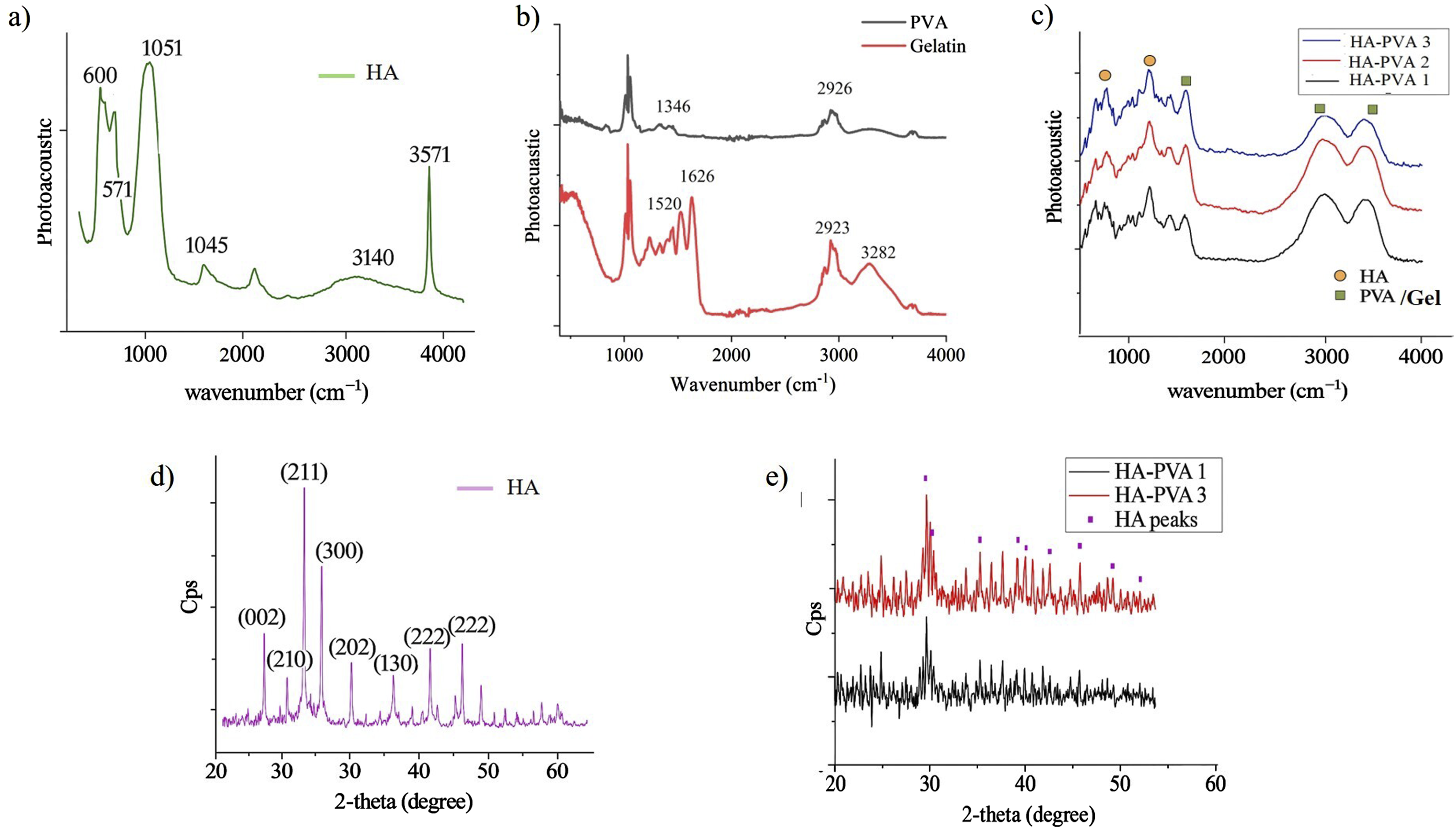
The FTIR spectra in Figure 1(c) confirmed the successful formation of composite scaffolds containing PVA, gelatin, and HA. The presence of HA was evidenced by characteristic peaks: an O-H stretching band at 3479 cm−1, two PO4−3 stretching bands at 1045 and 960 cm−1, and three PO4−3 bending bands at 601, 566, and 547 cm−1. 24 PVA-specific peaks were observed at 2912, 1411, and 830 cm−1, 26 while peaks at 3263, 1639 cm−1 were attributed to gelatin. 27 The intensity of HA-related peaks, particularly the O-H stretching and PO4−3 bands, increased proportionally with HA concentration in the scaffolds. In contrast, the PVA peaks at 3263 cm−1 and 2912 cm−1 maintained consistent intensities across all compositions. However, a slight decrease was observed for the PVA peak at 830 cm−1 as the PVA content decreased.
XRD
The XRD analysis of sintered HA powder demonstrated strong phase purity, with all diffraction peaks exhibiting precise alignment to the hexagonal HA reference (JCPDS 09-0432; Figure 1(d)). Characteristic peaks at 2θ = 25.7° (002), 27.9° (102), 28.7° (210), 31.6° (211), 32.0° (112), 32.7° (300), 33.9° (202), 39.6° (130), 46.5° (222), and 49.3° (213) confirmed the hexagonal P63/m symmetry. 28 Notably, sintering at 1100°C preserved phase stability, with no detectable formation of α- or β-tricalcium phosphate secondary phases. Crystallite size along the c-axis, calculated from the (002) diffraction plane using Scherrer analysis, measured 57.7 nm, consistent with nanostructured HA systems reported in prior studies. In contrast, XRD spectra of polymer-containing scaffolds showed reduced peak sharpness (FWHM increase of 18%–22%) and a 34% decrease in crystallinity index compared to pure HA powder (Figure 1(e)). This broadening effect correlates with polymer-induced inhibition of HA crystal growth, as observed in composite biomaterials, and suggests partial amorphisation at organic-inorganic interfaces.
SEM
SEM analysis showed cross-sectional architectures of HA-PVA scaffolds (compositions 1–3), demonstrating a hierarchical porous structure with interconnected macropores (100–300 µm) and surface micropores (Figure 2(a)). Quantitative pore size distributions for each composition are detailed in Supporting Information Figures S1–S3. In addition to pore size, the interconnectivity of the pores plays a crucial role in scaffold performance for tissue engineering, as it facilitates cell migration, nutrient diffusion, and waste removal. The interconnected architecture of scaffolds was qualitatively assessed using high-resolution SEM imaging (Figure 2). The images clearly revealed an open porous structure of HA-PVA 1 with channels linking adjacent pores, indicating good interconnectivity throughout the scaffold. Notably, HA-PVA 1 scaffolds exhibited optimal pore interconnectivity with average macropore diameters of 150 µm (Figure 2(a)). (a) SEM micrographs of freeze-dried scaffolds (Scale bar = 100 μm), (b) Compressive strength and porosity values of scaffolds, and (c) Stress-Strain curves for HA-PVA1, HA-PVA2 and HA-PVA3 scaffolds.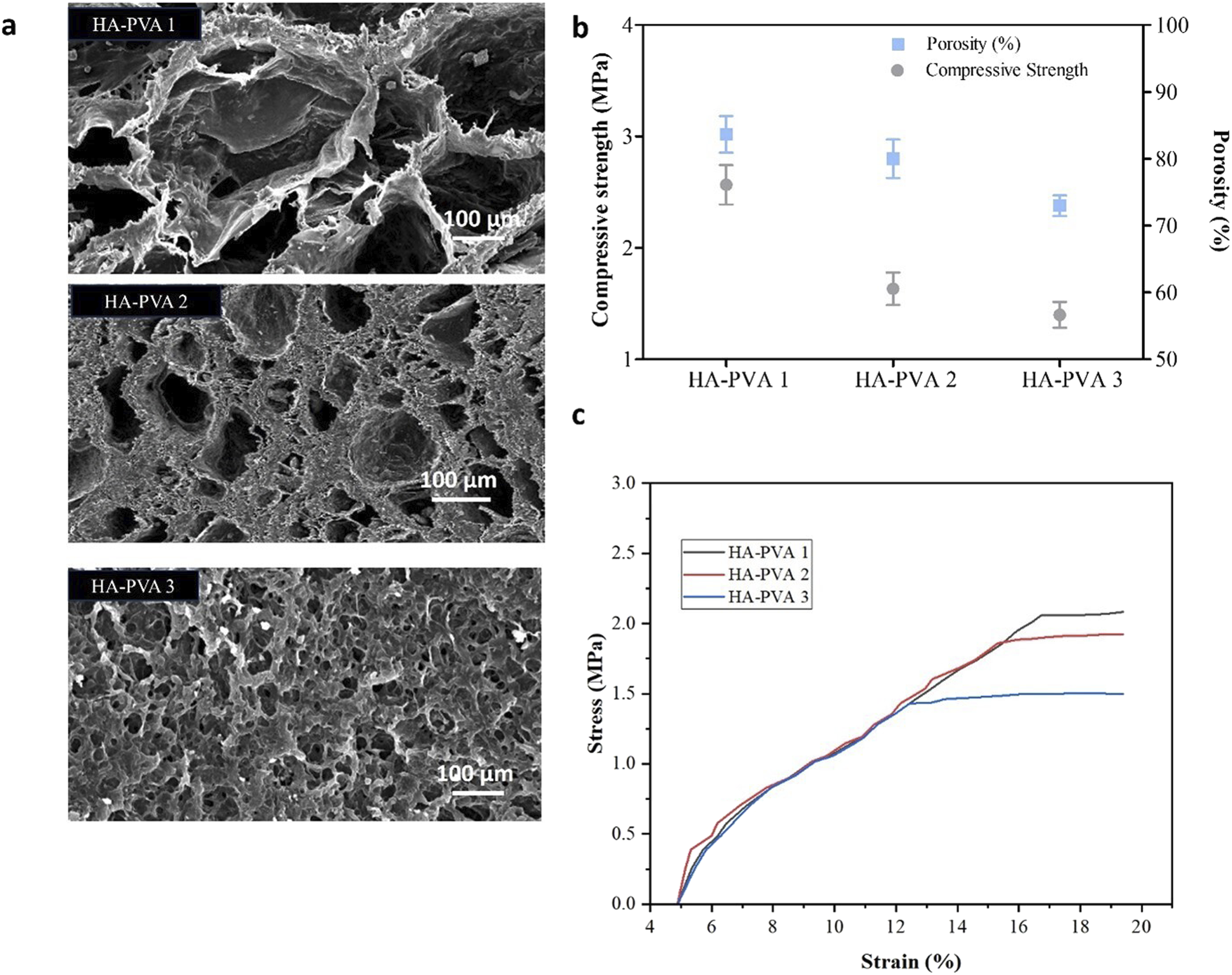
Progressive increases in ceramic content (HA-PVA 2 to HA-PVA 3) resulted in a proportional reduction in cumulative pore diameter and connectivity attributed to HA particles acting as nucleation sites that altered ice crystal growth dynamics during fabrication. 29 This phenomenon follows established trends in ceramic-reinforced polymer systems where particulate additives restrict crystal domain development. The engineered porosity profile offers three key functional advantages: (i) macropores (≥100 µm) facilitate vascular infiltration and nutrient diffusion; (ii) micropores (<20 µm) enhance drug payload capacity through the increased surface area, and (iii) gradient pore distribution promotes osteoblast attachment while maintaining mechanical integrity. 30
Porosity and mechanical properties
The scaffolds exhibited a porosity ranging from 73.8% to 84% (Table 1), which is sufficient to facilitate vascularisation and oxygen diffusion, thereby promoting osteogenesis. 31 Additionally, the compressive strength and Young’s modulus of all scaffold compositions confirm their suitability for spongy bone applications. Figure 2(b) demonstrates the relationship between porosity, compressive strength, and the composition of HA and PVA within the scaffolds. A decrease in the PVA-to-HA ratio from 2 to 0.5 resulted in a reduction in porosity from 83.6 ± 3.8% to 73.8 ± 2.1% and a corresponding decline in compressive strength from 2.675 ± 0.2 MPa to 1.504 ± 0.1 MPa.
In vitro tests
Biomineralisation
For a synthetic biomaterial to bond with living bone, it must promote the formation of bone-like apatite on its surface upon implantation, a process known as biomineralisation. This study evaluated the biomineralisation potential of HA/PVA/gelatin scaffolds through in vitro testing. Scaffold samples were incubated in SBF and formulated to replicate the ionic composition of human blood plasma. This approach enabled the assessment of the scaffolds capacity to induce mineralisation processes analogous to those occurring in natural bone formation.
FTIR and SEM analyses confirmed the successful in vitro mineralisation of the scaffolds. FTIR spectra exhibited characteristic peaks corresponding to mineral phases. Figure 3 provides a comparative analysis of HA/PVA scaffold spectra before and after immersion in SBF. On Day 0, the untreated scaffolds displayed distinct bands associated with PVA and HA within the scaffold matrix. The presence of deposited apatite was identified by phosphate bands in the spectral regions of 560–600 cm−1 and 960–1033 cm−1.
24
Initially, the intensity of these bands correlated with the HA content in the scaffold. After 7 days in SBF, the characteristic HA bands intensified, becoming more pronounced by Day 28, confirming the formation of HA on the scaffold surfaces. FTIR spectra of HA-PVA 1 and HA-PVA 3 scaffolds immersed in SBF for varying durations.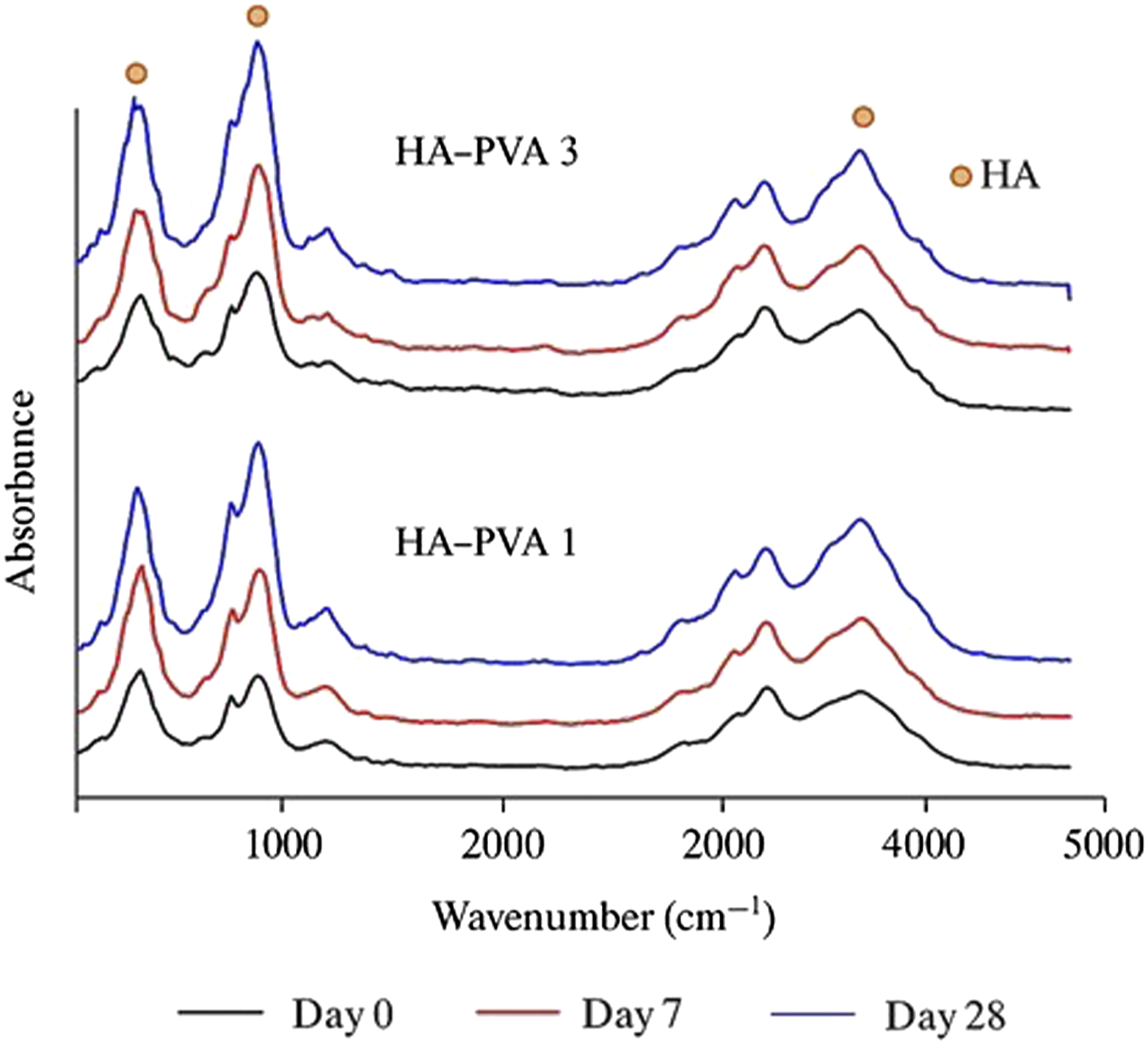
SEM imaging showed mineral deposition on the scaffold surfaces, demonstrating morphological changes over time following immersion in SBF. A thin apatite layer formed on the scaffold surfaces after 7 days, progressively thickening into a dense coating by Day 28. Concurrently, scaffold porosity decreased, and by Day 28, pores were entirely covered by a thick apatite layer (Figure 4). Comparisons of SEM images at 0 and 7 days showed more rapid mineralisation in the scaffold with high PVA content (HA-PVA 1) than in the scaffold with high HA content (HA-PVA 3). By Day 28, however, no statistically significant difference in apatite coverage was observed between these groups, indicating that all scaffolds supported substantial mineral deposition. The role of HA in this process arises from its intrinsic bioactivity: surface calcium and phosphate ions act as nucleation sites for apatite precipitation, which explains why all HA-containing scaffolds exhibited apatite formation irrespective of ratio. The more rapid mineralisation initially (at Day 7) observed in HA-PVA 1 can be attributed to its larger pore size and higher porosity, which facilitated ion transport and fluid penetration, thereby allowing the HA phase to interact more effectively with SBF.
32
The pores started filling with mineral clusters, indicating fast nucleation at Day 7. In contrast, higher HA loading (HA-PVA 3) provided more nucleation sites but reduced porosity, which may restrict transport pathways and delay early-stage deposition. The cluster deposition at Day 7 was evident but less extensive compared to HA-PVA 1 at same stage. However, by Day 28, significant mineral coverage was present for both scaffold types. Taken together, SEM and FTIR analyses confirmed that both composition and microstructure govern the mineralisation process, and that these scaffolds provide a bioactive environment conducive to bone bonding and growth. SEM images of scaffolds (a) HA-PVA 1 and (b) HA-PVA 3 at Day 0 (before immersion in SBF), Day 7, and Day 28 post-SBF immersion. (Scale bar = 100 μm).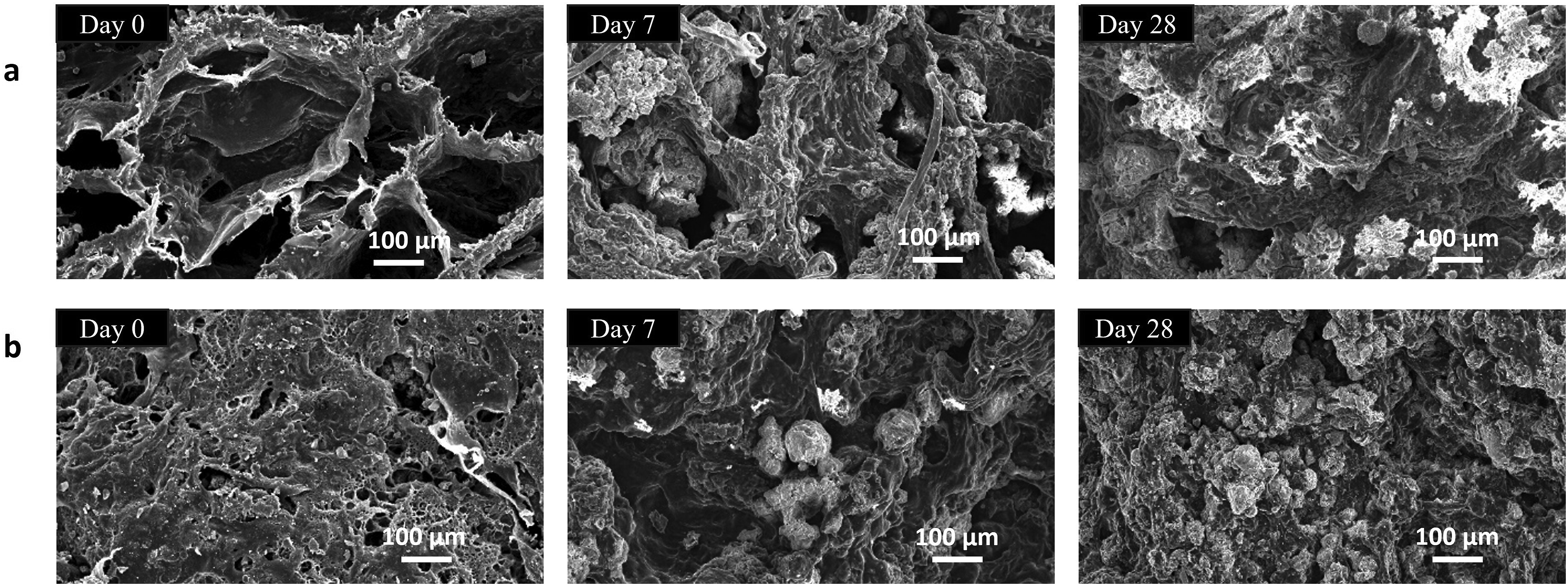
Swelling behaviour
The swelling study was conducted to evaluate the scaffolds’ fluid absorption capacity under physiological conditions. Swelling influences pore enlargement, facilitating nutrient diffusion and enhancing cellular infiltration and attachment, which are essential for tissue regeneration. The swelling behaviour of HA-PVA 1, HA-PVA 2, and HA-PVA 3 scaffolds (Figure 5(a)) indicated that all compositions reached their maximum swelling capacity by Day 7. This initial mass uptake was followed by a gradual reduction in swelling ratio over the subsequent 21 days, attributed to the rupture of polymer chains at higher swelling stages.
33
In terms of composition, swelling capacity decreased with increasing HA content. HA-PVA 1 exhibited the highest swelling ratio (9.0 ± 0.5) within the first 7 days, whereas HA-PVA 2 and HA-PVA 3 showed swelling ratios of 7.1 ± 0.5 and 3.6 ± 0.7, respectively. By Day 21, HA-PVA 1 retained a swelling ratio of 7.08 ± 1.0, while HA-PVA 2 and HA-PVA 3 exhibited reduced ratios of 5.1 ± 0.28 and 2.5 ± 0.5, respectively. Overall, the fluid uptake of HA-PVA 1 was significantly higher than that of HA-PVA 2 (p < 0.01) and HA-PVA 3 (p < 0.001). This greater swelling capacity can be attributed not only to its favourable material composition but also to its higher porosity compared to the other formulations. (a) Swelling ratios of the prepared scaffolds, (b) percentage degradation of the scaffolds, and (c) protein adsorption of the scaffolds in BSA at 37°C for 2, 4, 8, and 24 h. All data are presented as mean ± SD, n = 3. Statistical significance is indicated as *p < 0.05, **p < 0.01, and ***p < 0.001, compared to the HA-PVA 1 scaffolds.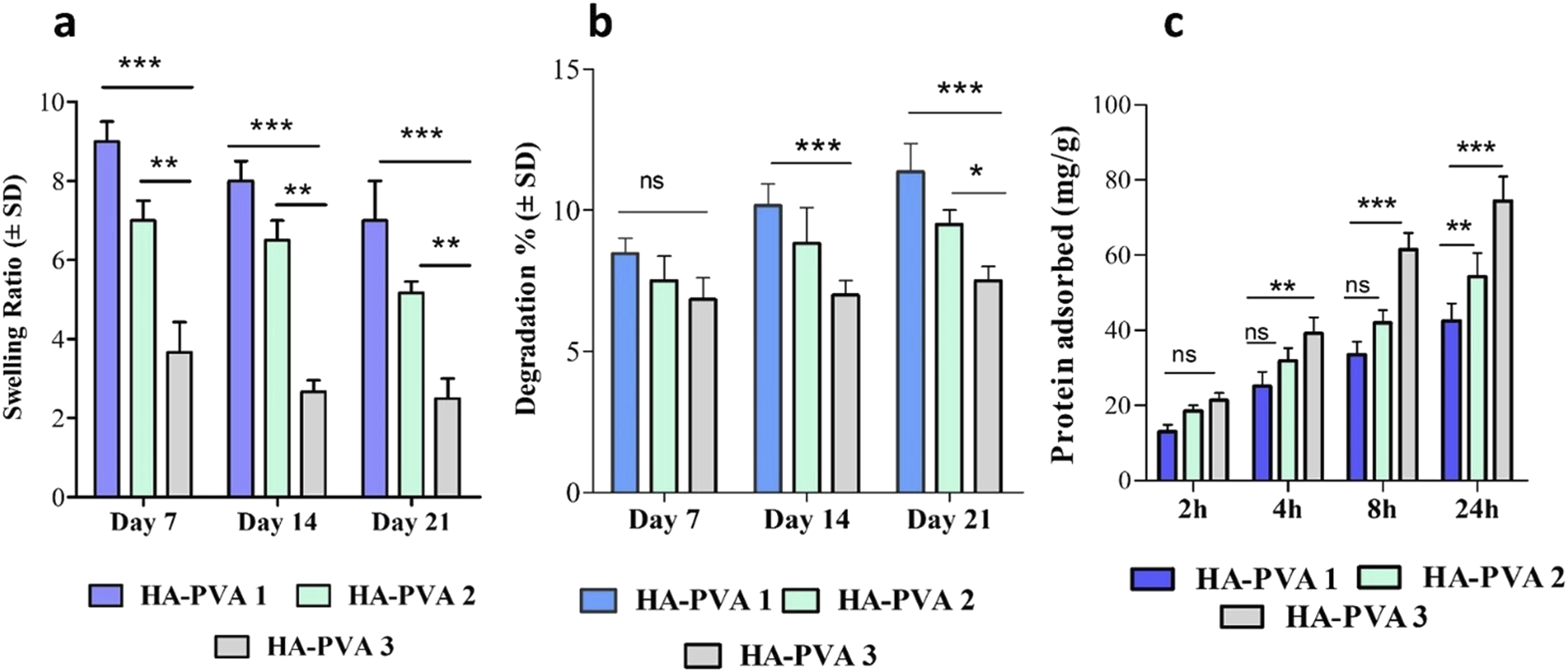
Biodegradation behaviour
The degradation analysis demonstrated that scaffolds with higher HA content degraded at a slower rate than those with lower HA content. Biodegradation tests were conducted in a lysozyme-containing PBS solution (Figure 5(b)). On Day 7, the degradation percentages for HA-PVA 1, HA-PVA 2, and HA-PVA 3 scaffolds were 8.4 ± 0.5%, 7.5 ± 0.1%, and 6.8 ± 0.05%, respectively. Thereafter, the scaffolds exhibited a gradual yet minimal weight loss over 21 days of incubation, indicating a slow degradation process.
By Day 21, the degradation percentages for HA-PVA 1, HA-PVA 2, and HA-PVA 3 scaffolds were 11.3%, 9.5%, and 7.5%, respectively (Figure 5(b)). These findings confirm that the biodegradation rate in an enzyme-containing PBS solution is influenced by the polymer-to-ceramic ratio within the scaffolds. Overall, all scaffold compositions demonstrated structural stability for up to 21 days, suggesting their potential to provide sustained support at the defect site during the bone regeneration process.
Protein adsorption
Protein adsorption on biomaterial surfaces from body fluids plays an important role in modulating cellular responses, including adhesion, proliferation, and differentiation, thereby influencing implant success. 34 The protein adsorption study demonstrated that the synthesised scaffolds effectively adsorbed proteins over time. After an initial 2-h incubation, the amount of protein adsorbed onto the HA-PVA 1, HA-PVA 2, and HA-PVA 3 scaffolds was 13.03 ± 1.8, 18.56 ± 1.4, and 21.3 ± 2.0 mg/g, respectively (Figure 5(c)). Extending the incubation period to 4 h increased protein adsorption to 25.1 ± 3.8, 31.9 ± 3.4, and 39.1 ± 4.3 mg/g, respectively. Further incubation for 8 and 24 h led to a significant increase in protein adsorption, reaching 42.4 ± 4.6, 54.3 ± 6.3, and 74.4 ± 6.5 mg/g, respectively.
All scaffolds exhibited a similar trend, with an initial rapid protein uptake followed by a gradual reduction in adsorption rate as binding sites became saturated. The HA-PVA 3 scaffolds adsorbed significantly more protein than the HA-PVA 1 scaffolds at 8 h and 24 h (p < 0.001) (Figure 5(c)). This improved adsorption is likely due to the increased presence of HA particles in the HA-PVA 3 scaffolds, which may alter surface properties such as chemical composition, electric charge, and morphology, thereby providing more active sites for protein interactions.35,36 The high inorganic HA content in the scaffolds (Figure 1(d)) further contributed to increased protein adsorption at prolonged incubation times. Overall, these findings suggest that all scaffolds exhibit favourable protein adsorption kinetics, supporting their potential to facilitate cell attachment and proliferation.
Haemolysis
Percentage haemolysis measurements of HA-PVA 1, HA-PVA 2, and HA-PVA 3 scaffolds.
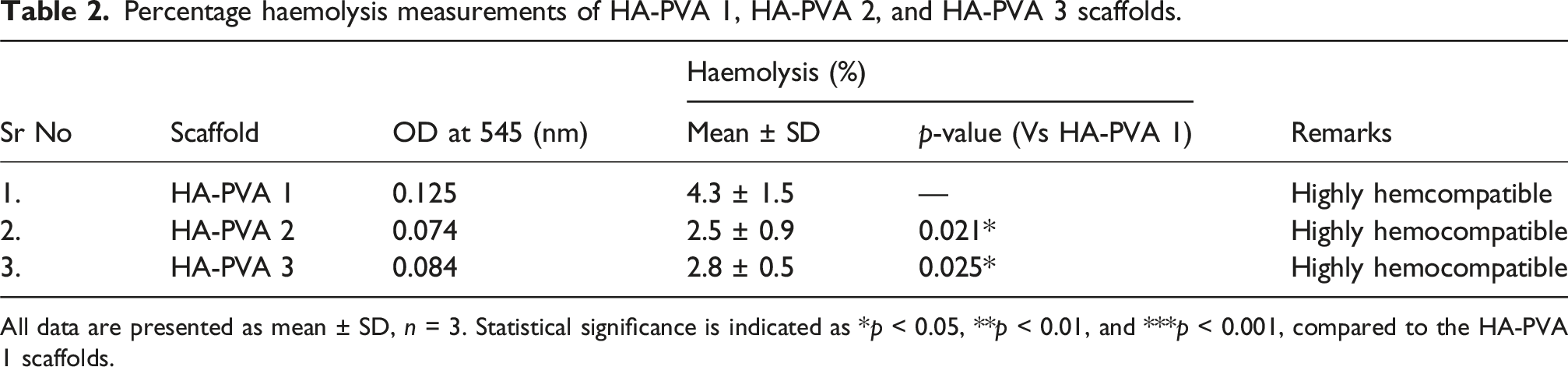
All data are presented as mean ± SD, n = 3. Statistical significance is indicated as *p < 0.05, **p < 0.01, and ***p < 0.001, compared to the HA-PVA 1 scaffolds.
Drug release and antibacterial activity
The release profile of the scaffolds exhibited a consistent trend: an initial rapid release followed by a sustained release phase. In vitro release data showed a pronounced burst release within the first 24 h (Figure 6(b)), attributed to the loosely adsorbed MOX on the scaffold surface. Following this initial burst, a sustained release was observed over the subsequent 72 h. (a) Bacterial inhibition zones of drug-loaded scaffolds against S. aureus. (b) Release profile of MOX from scaffolds over 72 h, expressed as percentage release. (c) Cytotoxicity assessment using the Alamar Blue assay. Data are presented as mean ± SD (n = 3), with statistical significance indicated as p ≤ 0.001 (***).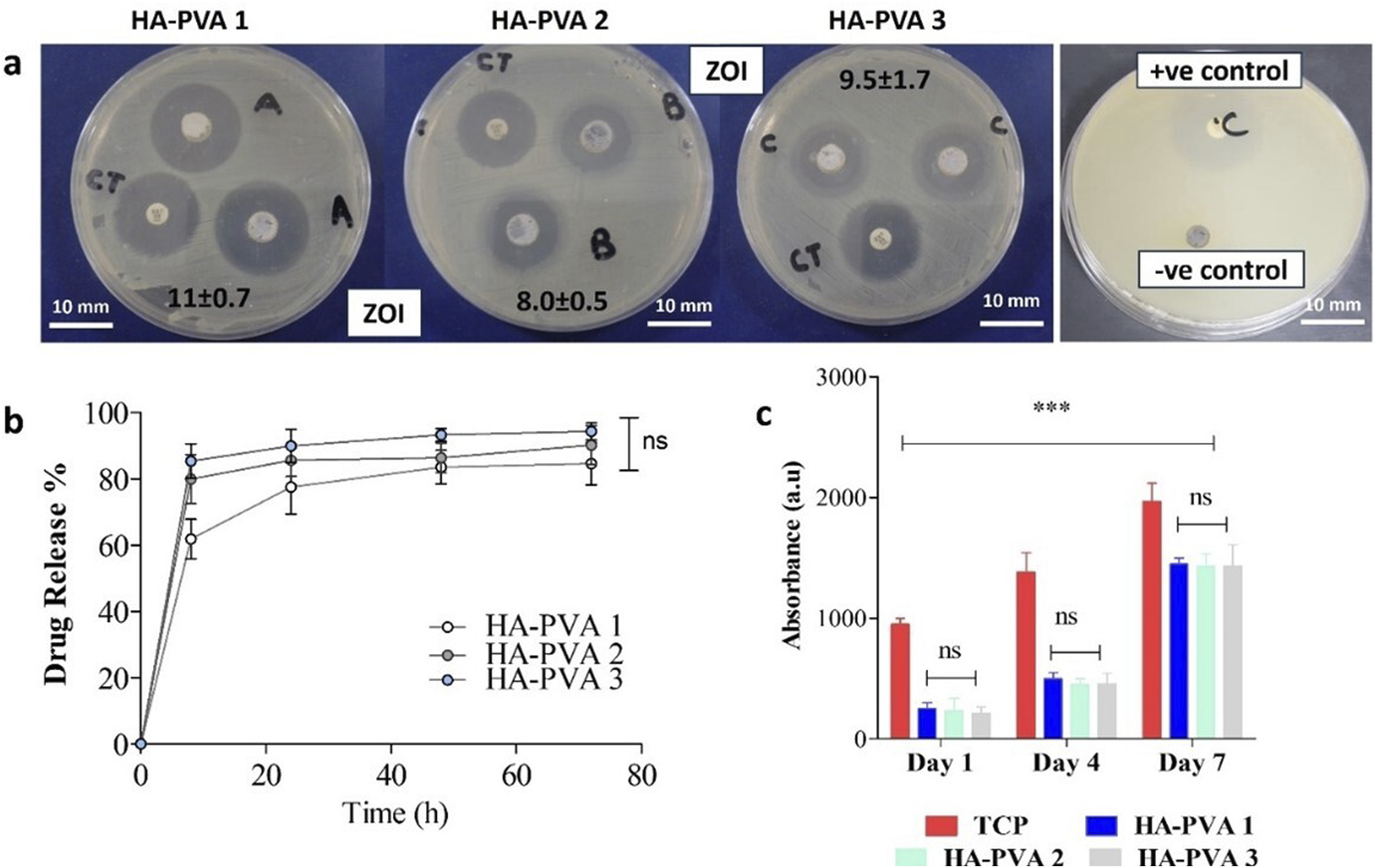
The observed release pattern is in line with expectations for chronic OM treatment, as suggested by previous studies. 38 The initial burst release is anticipated to rapidly elevate the local antibiotic concentration, facilitating the swift eradication of a significant proportion of pathogens. The subsequent gradual release phase helps to maintain an effective local concentration, thereby preventing bacterial colonisation and eliminating residual pathogens. This dual-phase release profile aligns with the requirements of antibacterial agents for chronic OM management, indicating its potential efficacy for treating infections in osseous defects.
The antibacterial efficacy of the MOX-loaded HA-PVA scaffolds (HA-PVA 1, HA-PVA 2, and HA-PVA 3) against S. aureus was evaluated by measuring the ZOI. Distinct transparent inhibition zones were observed around all MOX-loaded scaffolds. All drug-loaded scaffolds exhibited clear inhibition zones after 24 h of incubation, indicating excellent drug release performance (Figure 6(a)). Additionally, positive controls for each sample demonstrated antibacterial activity against S. aureus. Blank scaffolds (negative control) showed no antibacterial activity.
Cell viability
Cell viability, assessed using the Alamar Blue assay, demonstrated a time-dependent increase in cell proliferation across all three scaffold compositions (Figure 6(c)). On Day 7, the absorbance for all scaffolds was approximately 1500, compared to 250 on Day 1, indicating that hSMP cells remained viable and proliferated on the scaffolds. No significant differences in metabolic activity were observed between the scaffold compositions at any time point, suggesting that all tested scaffolds equally support cell growth and survival.
Discussion
This study examined the HA-PVA composite scaffolds’ physicochemical and biological performance, showcasing their promise as multipurpose platforms for localized drug delivery and bone tissue engineering. The effective integration of PVA, gelatin, and HA into the composite scaffolds was confirmed by the FTIR data. The broad O-H stretching at approximately 3263 cm−1, the C-H stretching at about 2912 cm−1, and the C-H bending and ring deformation at approximately 1411 and 830 cm−1 confirmed PVA and gelatin presence in composites that are consistent with previous observations. 39 Successful scaffold composition and homogeneity were further supported by the proportionate rise in HA-related peaks with increasing HA concentration. The well-organized porosity architecture of the HA-PVA scaffolds, essential for successful bone tissue regeneration, was confirmed by the SEM. The engineered gradient in pore size distribution within the HA-PVA scaffolds played a critical role in optimising their performance. The multi-scale porosity not only produces a physiologically favorable environment for cell proliferation but also enhances mechanical stability by even distribution of stress throughout the scaffold. The harmony between mechanical integrity and biological function emphasizes the synergistic benefits of hierarchical pore architectures, a design strategy increasingly recognised as essential for the successful bone regeneration. 40 In our study, scaffolds with a lower HA content (HA-PVA 1) showed higher compressive strength despite the fact that HA is inherently stiffer than PVA/gelatin. The interaction between composition and microstructure was reflected in the mechanical performance of HA-PVA scaffolds. The increased concentration of ceramic, which reduces the void space within the scaffold, was main reason for the decreased porosity as the HA content rises. However, strong hydrogen bond interactions between the hydroxyl groups of HA and the free hydroxyl groups in PVA, which improve interfacial bonding, were responsible for the high compressive strength seen in HA-PVA 1 with higher PVA concentrations.41,42 The lower PVA concentrations in HA-PVA 2 and HA-PVA 3 weakened these interactions, which lowers compressive strength. In addition, as demonstrated by SEM, the lower HA content in HA-PVA 1 resulted in larger pore sizes and higher overall porosity, which likely improved stress distribution and energy absorption 43 contributing to superior compressive strength of HA-PVA 1. This highlights that scaffolds mechanics are governed by HA content as well as by polymer–ceramic interactions and pore architecture.
The compressive stress-strain curves of scaffolds showed a distinctive multi-peak appearance that is typical of porous materials. 44 The curves exhibited an initial linear elastic zone followed by a collapse plateau (Figure 2(c)) with slight variations ascribed to the layer-by-layer collapse of the microstructure under compression. The compressive strength values within 0.2–4 MPa range (Table 1) are consistent with the mechanical benchmarks for cancellous (spongy) bone. 45
The scaffold porosity and surface properties have a major impact on in vitro bioactivity, as shown by the SEM observations of increasing apatite deposition on the scaffold surface. Increased mineralization exhibited by HA-PVA 1 post SBF immersion suggests that high porosity and interconnected pore networks facilitate ion exchange and fluid infiltration, which accelerates apatite nucleation and growth. 46 The more rapid and extensive apatite formation in the high-PVA scaffold, despite its lower HA content, highlights the significance of physical architecture of the scaffold in driving bioactivity. These findings, further supported by FTIR data, indicated the scaffolds potential for bone bonding and functional recovery by inducing apatite formation in SBF.
The swelling behavior demonstrated influence of porosity on scaffolds’ ability to absorb water. The substantial swelling in HA-PVA 1, particularly during the initial 7 days, reflected its higher porosity, which promoted better fluid penetration and retention—a relationship well-documented in scaffold design studies. 47 The subsequent decline in swelling ratios over 21 days may be attributed to polymer chain relaxation and partial degradation. Consistent with earlier studies, the degradation data demonstrated that the addition of HA successfully decreased the in vitro degradation rate of composite scaffolds. 48 This reduction is primarily attributed to the high crystallinity of HA, which slows resorption. The impact of HA crystallinity on its resorption rate is documented in several studies. For example, Klein et al. 49 found that highly crystalline HA did not exhibit degradation in a rabbit model until 9 months after implantation, while in a beagle alveolar bone model, poorly crystalline HA was considerably resorbed within 12 weeks. 50 The findings of the haemolysis assay showed that produced scaffolds exhibited haemolysis rates below the 5% threshold established by ISO 10993-4 for hemocompatibility, suggesting their safety to contact with blood. 51
The observed increase in cell proliferation over time indicated favorable scaffold–cell interactions in line with literature reporting that porous biomaterial scaffolds encourage cellular infiltration and nutrient exchange, hence increasing metabolic activity over time.52,53 The uniform metabolic activity observed in all scaffold types indicated that the biointerface with hSMP cells was not negatively affected by HA and PVA ratio variations. Instead, the porous architecture likely played overriding role by providing sufficient surface area, oxygen permeability, and nutrient exchange pathways which are the key factors for facilitating cell viability and proliferation.
The release profile further provides insight into the drug-binding mechanism within the scaffold. The initial burst phase indicated poorly adsorbed drug molecules to the scaffold surface, enabling quick desorption. 54 The sustained release phase, on the other hand, indicated diffusion-controlled release from the scaffold’s porous structure and a strong encapsulation of the antibiotic within the scaffold matrix. This dual-phase mechanism is advantageous for antimicrobial treatments because it ensures an early therapeutic action followed by prolonged drug availability, reducing the possibility of bacterial regrowth and improving treatment outcomes. 55 HA in the composite scaffolds played a dual role relevant to osteomyelitis management and bone regeneration. As the mineral phase of bone, HA is highly biocompatible and osteoconductive, providing nucleation sites for apatite deposition and promoting direct bonding with host tissue. This property supports osteointegration and guides new bone formation across the scaffold pores. By filling bone defects, it helps reduce spaces favorable for bacterial growth, thereby supporting osteomyelitis treatment. In addition, the porous HA contributes to drug adsorption and sustained release, as demonstrated by the controlled delivery of moxifloxacin making it particularly valuable in scaffolds designed for osteomyelitis treatment. Several previous studies have reported this role of HA in scaffolds for osteomyelitis management and bone repair.56,57
Overall, the structural, mechanical, and biological characteristics of the HA-PVA scaffolds demonstrated their potential as multifunctional biomaterials for bone tissue regeneration and localised drug delivery. Although all compositions of prepared scaffolds supported bioactivity, hemocompatibility and drug release, HA-PVA 1 (3:6:1) demonstrated the best overall trade-off between high interconnectivity, acceptable strength, and rapid mineralisation. To improve the ratios more methodically, follow-up research will employ a structured design-of-experiments approach.
Conclusions
In the present study, scaffolds designed to promote bone formation while concurrently delivering therapeutics for the treatment of bone infections, such as OM, were successfully developed. The desired pore size range (100–300 μm), critical for effective proliferation, was achieved by adjusting the weight percentages of HA and polymers during scaffold fabrication. The dual-pore size architecture of the scaffolds facilitated the controlled release of therapeutic agents. The medicated scaffolds exhibited an initial burst release of the drug, followed by a sustained release of MOX over an additional 72 h. Significant reductions in S. aureus colony counts were observed following scaffold incubation. The composite scaffolds demonstrated significant mineralisation in SBF and enhanced the proliferation of hSMP cells. The scaffolds also exhibited adequate mechanical properties, along with favourable in vitro bioactivity and biocompatibility. These findings underscore the potential of the scaffolds for local intra-osseous delivery of antimicrobials to treat OM, while simultaneously supporting bone regeneration.
Supplemental Material
Supplemental Material - Multifunctional biopolymer-hydroxyapatite composite scaffolds for antibiotic delivery in osteomyelitis treatment and bone regeneration
Supplemental Material for Multifunctional biopolymer-hydroxyapatite composite scaffolds for antibiotic delivery in osteomyelitis treatment and bone regeneration by Tehseen Riaz, Anila Asif, Rabia Zeeshan, Tanya J. Levingstone, Faiza Sharif and Nicholas Dunne in Journal of Biomaterials Applications
Footnotes
Acknowledgements
T.R. acknowledges technical assistance in blood compatibility tests and laboratory protocols provided by Dr. Ramla Shahid, COMSATS University Islamabad, Lahore campus.
Author contributions
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the HEC Pakistan (Higher Education Commission, Pakistan) under the grant number 20-1841/NRPU/R&D/HEC/105201.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The data that supports the findings of this study are available from the corresponding authors upon request.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.