
Abstract
Periodic reanalysis of genomic data plays a pivotal role in refining variant interpretation and resolving previously undiagnosed cases, particularly in the context of rare diseases. We report a female patient presenting with global developmental delay, drug-resistant epilepsy, optic atrophy, congenital heart defects, and craniofacial dysmorphism. An initially deprioritized heterozygous NOTCH1 variant (NM_017617.5: c.4787T>C; p.Leu1596Pro), previously associated with isolated cardiac phenotypes, was later reclassified as likely pathogenic following annual reanalysis through our institutional pipeline. Trio-based whole exome sequencing confirmed the variant's de novo origin, and emerging literature published in 2024 expanded the phenotypic spectrum of NOTCH1-related disorders to include neurologic, ocular, musculoskeletal, craniofacial, and integumentary features—closely mirroring the patient's presentation. This diagnostic refinement enabled tailored clinical management and informed genetic counseling. This case underscores the clinical utility of systematic genomic reanalysis in rare disease diagnostics, where evolving knowledge enables reclassification of previously uncertain variants. Moreover, this case adds to the growing body of evidence, broadening the recognized clinical and molecular landscape of NOTCH1-related disorders.
NOTCH1 gene encodes a transmembrane receptor involved in the Notch signaling pathway, playing a crucial role in endothelial-to-mesenchymal transition (EndMT) during embryonic development.1,2 Pathogenic variants in NOTCH1 are primarily associated with congenital heart defects, particularly aortic valve abnormalities.3,4 However, recent studies suggest that NOTCH1-related disorders may also encompass extracardiac manifestations, including neurodevelopmental, craniofacial, and ocular anomalies.5–7
In this report, we describe a patient with multisystem involvement and previously unexplained neurodevelopmental delay whose diagnosis was clarified through our annual reanalysis pipeline at ACURARE (Acibadem University Rare Diseases and Orphan Drugs Research Center). The NOTCH1 variant that was initially deprioritized was later classified as likely pathogenic (LP), based on new evidence and segregation analysis. As genomic diagnostics become more integrated into clinical practice, the utility of periodic reanalysis of sequencing data has gained increasing recognition. This approach allows the incorporation of updated phenotypic information and newly published genotype-phenotype correlations, enhancing diagnostic accuracy for unresolved cases. This case highlights the evolving phenotypic spectrum of NOTCH1-related disorders and the clinical utility of systematic variant reinterpretation.
Case Presentation
A 20-month-old female was referred to our pediatric genetics clinic due to global developmental delay and congenital heart defects. The patient was born to a 31-year-old mother (G1P1) at 39 weeks of gestation via elective cesarean section following a pregnancy complicated by polyhydramnios. Her birth weight was 3280 g (48th percentile) and the length was 50 cm (61st percentile). Soon after birth, echocardiographic evaluation revealed a moderate-sized ventricular septal defect and a secundum atrial septal defect. Developmental concerns became evident around 6 months of age, including poor visual tracking, absence of rolling or sitting, and delayed fine motor milestones. She began physical therapy following the pediatric neurology consultation, which led to partial motor improvement. However, developmental delays persisted, particularly in gross motor and communication domains. EEG demonstrated generalized spike-and-wave discharges that later localized to the left hemisphere, prompting treatment with levetiracetam.
By early 2021, she developed drug-resistant epilepsy that showed partial responsiveness to synacthen and adjunctive treatment with valproate and clonazepam. Brain magnetic resonance imaging revealed diffuse hypomyelination, thinning of the corpus callosum, progressive cerebral and cerebellar atrophy, optic atrophy, and chronic intracranial calcifications. Ophthalmologic evaluation revealed tortuous retinal vessels and a suspected arteriovenous malformation, for which laser therapy was applied. Facial dysmorphism included thin arched eyebrows, a prominent philtrum, hypertrichosis, and small hands and feet. Neurologic examination revealed axial hypotonia, peripheral spasticity, and brisk deep tendon reflexes (Table 1).
Clinical Summary of the Proband.

Pedigree analysis (Figure 1A) revealed no significant familial disease history, and the parents were nonconsanguineous. A chromosomal microarray analysis was performed to explore potential genetic causes, and the results were negative. Whole exome sequencing (WES) conducted at an external laboratory identified a heterozygous variant in the COL4A3BP gene (NM_001130105.1: c.157T>C; p.Trp53Arg), inherited from the unaffected father. Given the lack of segregation with disease and insufficient phenotypic overlap, this variant was classified as a variant of uncertain significance (VUS) 8 and considered noncontributory.
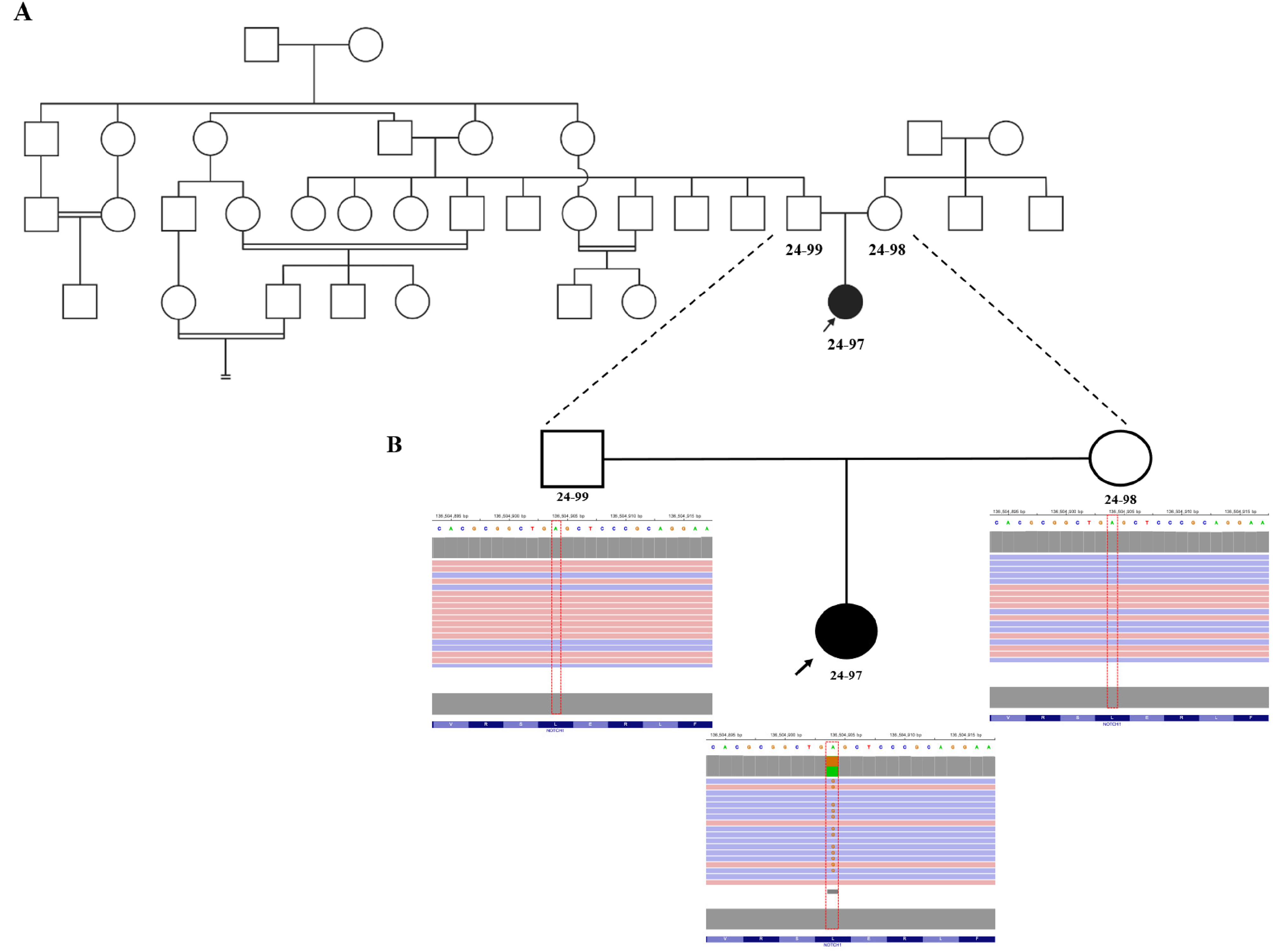
(A) Pedigree of the family and Integrative Genomics Viewer (IGV) snapshots demonstrating segregation of the NOTCH1 variant at chr9:136,504,904A>G (NM_017617.5:c.4787T>C; p.Leu1596Pro). (B) The proband (24-97) is heterozygous for the variant with high-quality sequencing metrics (total depth = 207 reads; allele balance = 0.498). Both parents are homozygous reference (wild type): father (24-99, depth = 230 reads) and mother (24-98, depth = 280 reads), confirming the de novo origin of the variant. High read depth (>200×) and appropriate allele ratios across all samples support the reliability of the variant call.
Because of the absence of a definitive molecular diagnosis and the patient's evolving phenotype, she was enrolled in ACURARE's annual genetic reanalysis program. Although a heterozygous variant in the NOTCH1 gene (NM_017617.5:c.4787T>C; p.Leu1596Pro; NC_000009.12:g.136504904A>G) had been initially detected during solo WES in 2021, it was filtered out because of its primary association with isolated cardiac defects, which did not fully explain the patient's complex multisystem phenotype. At that time, no supporting literature linked this variant to extracardiac features such as neurologic or craniofacial anomalies.
In 2023, reanalysis was prompted by the family's inquiry regarding pregnancy planning and genetic counseling, which triggered comprehensive reassessment of the genomic data through our institutional protocol (detailed methodology in Supplementary File 1). The trio-based reanalysis of the original WES data led to re-evaluation of this NOTCH1 variant. The variant is a single-base substitution located in the middle of exon 26 within a 34-exon transcript, resulting in a leucine-to-proline substitution at position 1596 in the NOTCH1 protein. This substitution occurs within a conserved EGF-like repeat domain—a known mutational hotspot relevant to receptor function. Trio analysis confirmed that the variant had arisen de novo. The variant was absent in gnomAD v4, 9 the 1000 Genomes Project, 10 ClinVar, 11 and HGMD databases, 12 and was predicted to be deleterious by multiple in silico prediction tools. Copy number variant analysis from WES data yielded negative results, and no additional causative variants were identified in genes previously associated with the patient's clinical presentation.
A comprehensive literature review identified a newly published 2024 study establishing a link between NOTCH1 variants and a broader range of phenotypes, including neurodevelopmental delay, optic atrophy, cerebral atrophy, and craniofacial dysmorphism—closely mirroring our patient's presentation. 7 This convergence of updated clinical and genetic evidence strengthened the variant's potential diagnostic significance. Following ACMG/AMP guidelines, 8 the variant was classified as LP based on the following criteria: PS2, PM2, and PP3 (Supplementary File 1, Supplementary Table 1).
Discussion
Pathogenic variants in NOTCH1 affect a highly conserved transmembrane receptor involved in the Notch signaling pathway, which plays a pivotal role in embryogenesis, vascular development, ocular development, and cell fate determination. Although it has been classically associated with autosomal dominant aortic valve disease and Adams-Oliver syndrome,5,6 emerging evidence suggests a broader phenotypic spectrum encompassing complex congenital heart disease, optic nerve abnormalities, and various extracardiac manifestations, including neurodevelopmental involvement. 7
Our case expands this phenotypic spectrum and underscores the significance of ophthalmologic findings—specifically optic nerve atrophy—which remain rarely reported in association with NOTCH1 variants. 7 Given the established role of NOTCH1 haploinsufficiency in neurovascular development, optic atrophy in our patient may represent either an underrecognized primary manifestation or a secondary consequence of impaired vascular remodeling. Nonetheless, the possibility of an additional contributing mechanism cannot be excluded, and further cases will be required to determine whether optic atrophy constitutes a reproducible component of the NOTCH1 phenotypic spectrum.
Although a few previously published cases noted similar ocular features, these were often accompanied by limited neurodevelopmental characterization or milder systemic involvement.5,6 In contrast, our patient exhibited a syndromic presentation characterized by global developmental delay, epilepsy, craniofacial dysmorphism, and progressive visual impairment. This constellation of features strengthens the emerging concept of a multisystem NOTCH1-related disorder and contributes to the continued refinement of its phenotypic landscape.
A critical turning point in our patient's diagnostic journey was the publication by Stanley et al 7 in 2024, which expanded the known phenotypic spectrum of NOTCH1-related conditions, particularly in relation to neurologic and ocular manifestations—features that had previously been under-recognized. Although the NOTCH1 variant (p.Leu1596Pro) had been identified during initial solo WES in 2021, it was deprioritized because of its primary association with isolated cardiac defects, which did not adequately explain our patient's complex multisystem phenotype.
The identified p.Leu1596Pro variant affects a conserved EGF-like repeat within the extracellular domain of NOTCH1. Accumulating genetic and functional evidence indicates that pathogenic NOTCH1 variants in EGF-like repeat domains result in loss of function through haploinsufficiency rather than gain-of-function or dominant-negative mechanisms. Southgate et al 13 demonstrated that NOTCH1 haploinsufficiency leads to reduced receptor expression and downregulation of downstream Notch targets, including HEY1 and HES1, establishing reduced signaling output as the primary disease mechanism. Consistently, Stittrich et al 6 showed that heterozygous NOTCH1 mutations cause Adams-Oliver syndrome through insufficient Notch signaling rather than aberrant receptor activity 6 . In a comprehensive cohort analysis, Stanley et al7 further confirmed that both truncating variants and whole-gene deletions produce overlapping phenotypes, providing strong evidence against dominant-negative effects. In addition, missense variants affecting EGF-like repeats have been shown to impair receptor folding, endoplasmic reticulum trafficking, and S1 cleavage, ultimately reducing the availability of functional receptor at the cell surface. 14 On this basis, the p.Leu1596Pro variant is strongly predicted to act through a loss-of-function mechanism consistent with the established haploinsufficiency model for NOTCH1-associated disorders, providing a unifying explanation for the patient's multisystem phenotype.
The periodic re-evaluation of genomic data through our annual reanalysis protocol allowed for a renewed assessment of this variant's clinical relevance. Incorporating the newly published evidence from Stanley et al,7 along with the confirmed de novo status of the variant through trio analysis, enabled its reclassification as LP, leading to a precise diagnosis and more informed clinical management. This diagnostic breakthrough shows the vital importance of systematic reanalysis in the context of rare and undiagnosed conditions, where scientific advancements continuously reshape genotype-phenotype interpretations. Recognition of haploinsufficiency as the underlying disease mechanism further provides a unifying explanation for the multisystem involvement observed in our patient, including neurologic and ophthalmologic involvement, in line with the expanded neurologic phenotype now reported for pathogenic NOTCH1 variants.
Nevertheless, several challenges persist in the field of genetic diagnostics. Interpretation of variants is often complicated by incomplete phenotypic data, discrepancies among genomic databases, and the complexity inherent to rare disease presentations.8,15 Additionally, the unavailability of parental samples can hinder the confirmation of segregation, which is essential for accurate variant classification. Our case exemplifies how these barriers can be overcome through structured reanalysis programs that integrate evolving clinical and molecular evidence. Addressing remaining challenges requires continued collaboration, the implementation of standardized reanalysis protocols, and the expansion of publicly accessible variant repositories to support precise and consistent variant interpretation.
Conclusion
This case highlights the critical importance of periodic genetic reanalysis in clinical practice, particularly for rare or undiagnosed conditions in which evolving scientific knowledge refines genotype-phenotype correlations. Through systematic reanalysis, a previously deprioritized NOTCH1 variant was re-evaluated and reclassified as likely pathogenic, with trio analysis enabling segregation assessment and newly published literature providing essential phenotypic and mechanistic context. Recognition of haploinsufficiency as the underlying disease mechanism offers a unifying explanation for the patient's multisystem involvement and informs both clinical management and genetic counseling. As genomic medicine advances, the capacity to correlate genetic variants with clinical features continues to improve, leading to more accurate diagnoses and optimized patient care. Routine reanalysis ensures that patients benefit from the most current genetic insights, ultimately improving clinical outcomes.
Supplemental Material
sj-docx-1-jcn-10.1177_08830738261422860 - Supplemental material for Periodic Genetic Reanalysis Identifies a Novel De Novo NOTCH1 Variant: A Case Report
Supplemental material, sj-docx-1-jcn-10.1177_08830738261422860 for Periodic Genetic Reanalysis Identifies a Novel De Novo NOTCH1 Variant: A Case Report by Eylul Aydin, Aybike S. Bulut, Berkay Yildiz, Ulas Ozonur, Ozkan Ozdemir, Kaya Bilguvar, Ozden Hatirnaz Ng, Burak Tatli, Ozlem Akgun-Dogan and Yasemin Alanay in Journal of Child Neurology
Footnotes
Acknowledgements
We would like to thank the patient and her family for their contribution. Eylul Aydin is supported by the Acibadem University Kerem Aydinlar Foundation Doctoral Scholarship for Academic Excellence.
ORCID iDs
Author Contributions
Eylul Aydin contributed to conceptualization, genomic data reanalysis, variant interpretation, and manuscript writing. Aybike S. Bulut contributed to genomic data analysis, variant interpretation, and manuscript writing. Berkay Yildiz contributed to data collection, manuscript review, and editing. Ulas Ozonur contributed to data collection, manuscript review, and editing. Ozkan Ozdemir contributed to methodological supervision and manuscript review. Kaya Bilguvar contributed to methodological supervision and manuscript review. Ozden Hatirnaz Ng contributed to methodological supervision and manuscript review. Burak Tatli contributed to clinical evaluation and manuscript review. Yasemin Alanay contributed to clinical genetic assessment and manuscript review. Ozlem Akgun-Dogan contributed to conceptualization, supervised the study and critically revised the manuscript. All authors approved the final version of the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was a part of a project that was funded by Istanbul Kalkinma Ajansi (Istanbul Development Agency), Turkey under the project number of TR10/22/TNH/0001.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.