
Abstract
Purpose
Evaluation of the incidence and variability of ocular manifestations in children with neurofibromatosis type 1.
Methods
In this study, the files of 71 children aged 0-18 years with neurofibromatosis type 1 were retrospectively analyzed. Child age groups were categorized as 0-6, 7-12, and 13-18 years. In cycloplegic refractive examination, ≥−0.50 Diopter (D) values in spherical equivalents were recorded as myopia, ≥+2.0 D as hypermetropia, and ≥±1.0 D cylindrical values as astigmatism. Patients with a difference of ≥1 D in spherical or cylindrical equivalents between the 2 eyes were considered anisometropic. Amblyopia was defined as a best-corrected visual acuity ≤0.8 with Snellen chart and a difference of at least 2 lines between both eyes. The presence of 2 or more iris Lisch nodules (iris hamartoma) was considered positive.
Results
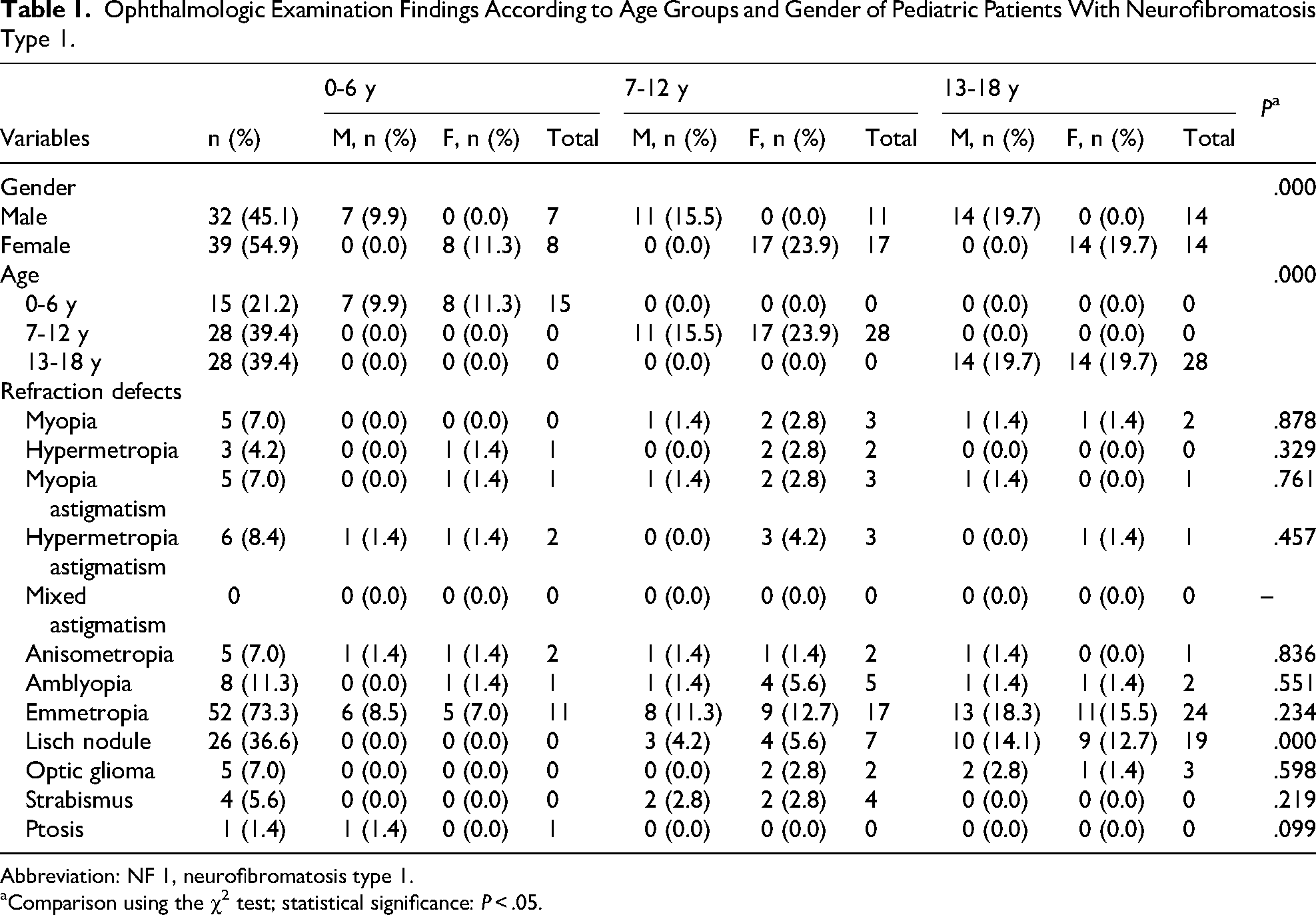
Of the 71 patients whose ocular findings were evaluated, 32 (45.1%) were boys and 39 (54.9%) were girls. According to age and gender, myopia (P = .878), hypermetropia (P = .329), myopia astigmatism (P = .761), hypermetropia astigmatism (P = .457), mixed astigmatism, anisometropia (P = .836), amblyopia (P = .551), emmetropia (P = .234), optic glioma (P = .598), strabismus (P = .219), and ptosis (P = .099) showed no significant difference (P > .05). A statistically significant difference was observed in the Lisch nodule, one of the ocular examination findings, according to age and gender (P < .05).
Conclusions
Pediatric patients with neurofibromatosis type 1, with common ocular manifestations, should undergo a comprehensive ophthalmologic examination. Early diagnosis and treatment are crucial for improving the clinical course of the disease and preserving vision.
Phakomatoses are a group of congenital disorders with genetic and multisystem involvement. It includes hamartomatous lesions of ectoderm origin. The involvement of the nervous system, eye, and skin is usually observed. It is also called a neuro-oculo-cutaneous syndrome because of the frequency of ocular involvement. Neurofibromatosis is a phakomatosis that primarily affects the cell growth of nervous tissue. It consists of 2 different genetic types and is characterized by the formation of neuroectodermal tumors in multiple organs in both types: neurofibromatosis type 1 and neurofibromatosis type 2. Neurofibromatosis type 1 is the most common phakomatosis, also known as von Recklinghausen disease. Its prevalence is 1/3000. It shows autosomal dominant inheritance. It is not related to gender, race or geographical status. It can be seen in all races, ethnicities, and genders. Spontaneous mutations cause 50% of the disease, and the other 50% is inherited.1–10 neurofibromatosis type 1 is associated with various ocular symptoms. The eye and ocular adnexa are frequently affected in patients with neurofibromatosis type 1. The most common ocular finding of neurofibromatosis type 1 is Lisch nodules in the iris. Other ocular findings include optic gliomas, orbital neurofibromas, congenital absence of sphenoid wing, congenital glaucoma, choroidal hamartomas, and retinal tumors. The clinical features of neurofibromatosis type 1 can be seen from birth, and the clinical picture becomes more prominent in childhood and adolescence with age. The distinctive feature of neurofibromatosis type 2 is the appearance of bilateral vestibular schwannomas. Ophthalmic findings, including optic glioma, Lisch nodules, orbital bone dysplasia, and periorbital plexiform neurofibroma, are some of the diagnostic criteria of neurofibromatosis type 1.3–5,7,11 Causes of vision loss in neurofibromatosis type 1 are frequently optic tract gliomas, orbital plexiform neurofibroma, and glaucoma.
The National Institutes of Health has developed diagnostic criteria for neurofibromatosis type 1. The presence of 2 or more of the following clinical features is diagnostic: 6 or more café au lait macules with a diameter of 5 mm in preadolescents and 15 mm in adults, 2 or more neurofibromas or 1 or more plexiform neurofibromas, 2 or more Lisch nodules, optic glioma, a prominent bone lesion, or a first-degree relative with neurofibromatosis type 1 findings. 8 In a study, it was reported that the order of presentation of clinical features was generally café au lait macules, axillary freckling, Lisch nodules, and neurofibromas. It has been reported that symptomatic optic glioma usually develops at the age of 3 years and characteristic bone lesions usually develop in the first year of life. This is important for diagnostic criteria in pediatric patients aged <8 years. 12
Ophthalmologists play a critical role in preventing visual loss and diagnosing and follow-up of the disease. This study aims to evaluate the incidence and variability of ocular manifestations in children with neurofibromatosis type 1.
Methods
In this study, 71 children (32 boys, 39 girls) aged 0-18 years with neurofibromatosis type 1 who were consulted from University Pediatric Neurology Clinic to Ophthalmology Clinic between January 2018 and November 2023 were retrospectively analyzed. All children in this study had a definite diagnosis of neurofibromatosis type 1, and at least 2 diagnostic criteria were present. Ethical approval was obtained with the number 2024/6551 from the Ethics Committee of University Faculty of Medicine. This study was performed in accordance with the Declaration of Helsinki.
The age groups of children were categorized as 0-6, 7-12, and 13-18 years. Visual acuity was measured using Snellen chart, and the best-corrected visual acuity was recorded using the logMAR scale. Children were instilled 2 drops of 1% cyclopentolate in each eye at 10-minute intervals, and cycloplegic refraction values were measured 40 minutes later when the pupil was dilated. In cycloplegic refraction examination, values of −0.50 Dioptre (D) and higher in spherical equivalents were recorded as myopia, ≥+2.0 D as hypermetropia, and cylindrical values of ≥±1.0 D and higher were recorded as astigmatism. Patients with a difference of ≥1 D in spherical or cylindrical equivalents between the 2 eyes were considered anisometropic. Amblyopia was defined as a best-corrected visual acuity ≤0.8 with Snellen chart and a difference of at least 2 lines between both eyes. Detailed ophthalmologic examination, including slit-lamp biomicroscopy, anterior segment examination, intraocular pressure measurement with Goldmann tonometry, pupillary reflexes, ocular motility, fundus examination with +90 D lens after pharmacologic dilatation, and eyelid examination was performed. The presence of 2 or more iris Lisch nodules (iris hamartoma) was considered positive. Optic pathways were visualized by eye and brain magnetic resonance imaging (MRI).
Statistical Analysis
Data were analyzed using SPSS, version 27.0 software (Statistical Package for the Social Sciences). Descriptive statistical methods (mean, standard deviation, percentage) were used to evaluate the data. The χ2 test was used to analyze all categorical variables according to age and gender groups. Statistical significance was accepted at a 5% alpha level.
Results
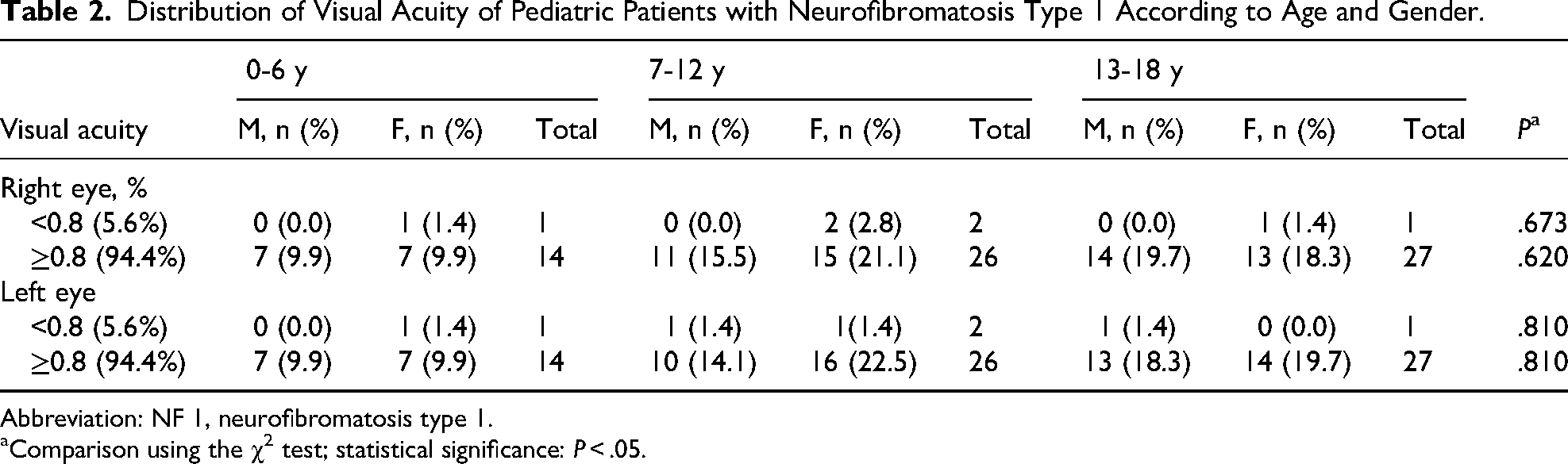
In this study, the files of 71 pediatric patients aged 0-18 years diagnosed with neurofibromatosis type 1 were retrospectively analyzed. Thirty-two (45.1%) of the patients were boys, and 39 (54.9%) were girls. The mean age of boys and girls was 11.7 ± 5.3 and 11.6 ± 4.5 years, respectively. Gender and age variables differed significantly according to age and gender (P < .05). Male (female) children aged 13-18 years are more than male (female) children aged 0-6 years. Children aged 7-12 and 13-18 years are more than children aged 0-6 years. Myopia (P = .878), hypermetropia (P = .329), myopia astigmatism (P = .761), hypermetropia astigmatism (P = .457), mixed astigmatism, anisometropia (P = .836), amblyopia (P = .551), emmetropia (P = .234), optic glioma (P = .598), strabismus (P = .219), and ptosis (P = .099) showed no significant difference (P > .05). There was a significant difference in Lisch nodule, which is one of the findings of ocular examination according to age and gender (P < .05). Lisch nodules were observed more in boys and girls aged 13-18 years than in children aged 7-12 years. Because no patient had mixed astigmatism, χ2 analysis did not yield a P value (Table 1 and Figure 1). There was no difference in visual acuity for the right and left eye according to age groups and gender (P > .05). Visual acuity in the right eye of 1 girl (1.4%) in the 0-6-year age group, 2 girls (2.8%) in the 7-12-year age group, and 1 girl (1.4%) in the 13-18-year age group was <0.8 (P = .673), and visual acuity in the left eye of 1 girl (1.4%) in the 0-6 age group, 1 girl (1.4%) and 1 boy (1.4%) in the 7-12-year age group, and 1 boy (1.4%) in the 13-18-year age group was <0.8 (P = .810). The findings showed right eye visual acuity ≥0.8 (P = .620) in 7 boys (9.9%) and 7 girls (9.9%) in the 0-6-year age group, 11 boys (15.5%) and 15 girls (21.1%) in the 7-12-year age group, and 14 boys (19.7%) and 13 girls (18.3%) in the 13-18-year age group. Also, 7 boys (9.9%) and 7 girls (9.9%) in the 0-6-year age group, 10 boys (14.1%) and 16 girls (22.5%) in the 7-12-year age group, and 13 boys (18.3%) and 14 girls (19.7%) in the 13-18-year age group had left eye visual acuity ≥0.8 (P = .810) (Table 2 and Figure 2).
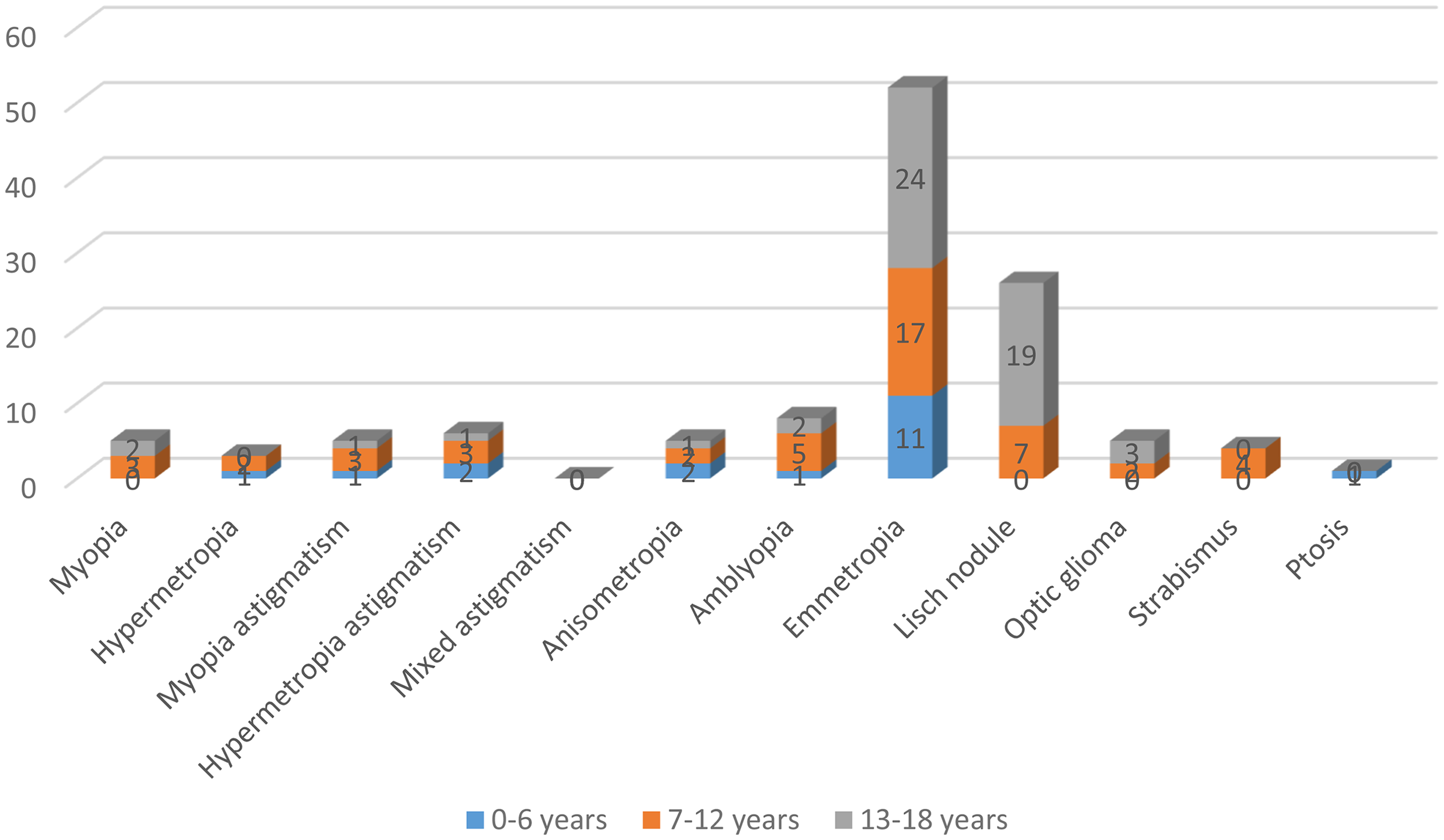
Ophthalmological Examination Findings According to Age Groups of Pediatric Patients with Neurofibromatosis Type 1. The Majority of Children in All Age Groups are Emmetropic, and the Most Common Ophthalmological Examination Finding is Lisch Nodule.
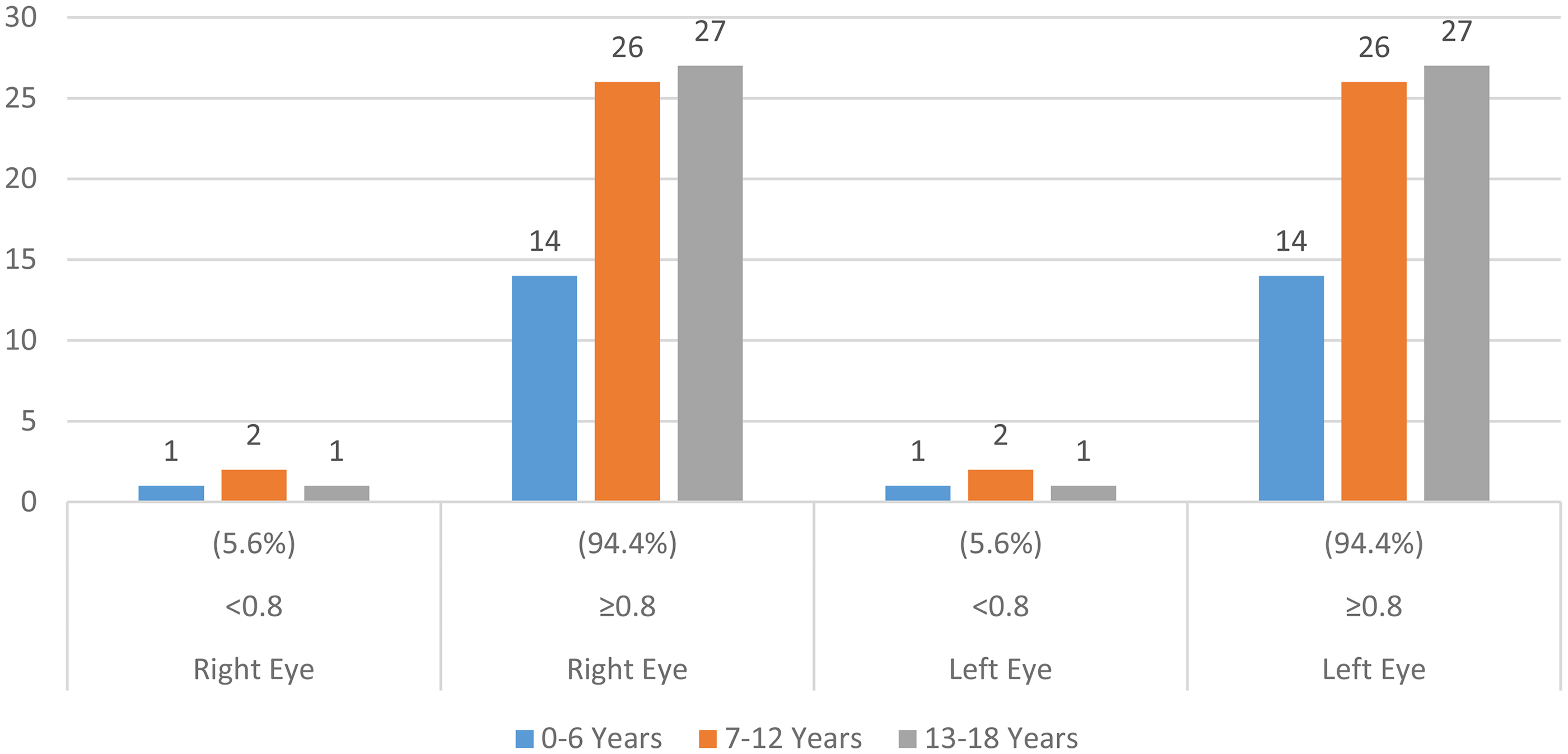
Findings of Visual Acuity of Pediatric Patients with Neurofibromatosis Type 1 According to Age. Visual Acuity is Most Commonly 0.8 or Higher in Both Eyes in All Age Groups (0-6 Years, 7-12 Years, 13-18 Years).
Ophthalmologic Examination Findings According to Age Groups and Gender of Pediatric Patients With Neurofibromatosis Type 1.
Abbreviation: NF 1, neurofibromatosis type 1.
Comparison using the χ2 test; statistical significance: P < .05.
Distribution of Visual Acuity of Pediatric Patients with Neurofibromatosis Type 1 According to Age and Gender.
Abbreviation: NF 1, neurofibromatosis type 1.
Comparison using the χ2 test; statistical significance: P < .05.
Discussion
Neurofibromatosis is a neurocutaneous syndrome that primarily affects the cell growth of neural tissue. It consists of 2 different genetic types, and both types are characterized by the formation of neuroectodermal tumors in more than 1 organ.3,13 Genetic tests are crucial in diagnosing uncertain cases or when prenatal genetic diagnosis is requested. Neurofibromin, the neurofibromatosis type 1 gene protein product, is a tumor suppressor especially in neurons, glial cells, Schwann cells and early stages of melanocyte development. Functional loss of neurofibromin leads to uncontrolled cell proliferation and thus increases the risk of neoplasm. 3
Ophthalmic findings, including optic glioma, Lisch nodules, orbital bone dysplasia, and periorbital plexiform neurofibroma, constitute an important part of the diagnostic criteria of neurofibromatosis type 1. The clinical features of neurofibromatosis are present at birth and become apparent during childhood and adolescence. The fact that neurofibromas are commonly observed in adolescence suggests that these tumors may be stimulated by changes in hormones.7,8
Lisch nodules are the most common ocular finding. They are usually specific for neurofibromatosis type 1 and are primarily bilateral. They are benign, yellow to brown colored, dome-shaped protrusions and histologically melanocytic hamartomas. They tend to cluster in the lower half of the iris, probably because of the sunlight-shielding effects of the upper eyelid. Lisch nodules are usually not present at birth, and their incidence gradually increases with age. They are not expected to be seen before 2 years of age. The absence of Lisch nodules in children does not rule out the diagnosis of neurofibromatosis type 1.3,5,14 Kordic et al, 13 in a study performed in 132 pediatric patients, found Lisch nodules with a rate of 78%. In their study, Zimmermann et al 15 reported that 66% of 119 of pediatric patients had Lisch nodules. Çarman et al 16 found Lisch nodules in 49 pediatric patients with a rate of 40.8%. In this study, Lisch nodules were recorded with a rate of 36.6% in all children, with the highest rate in the 13-18-year age group (Figure 3).
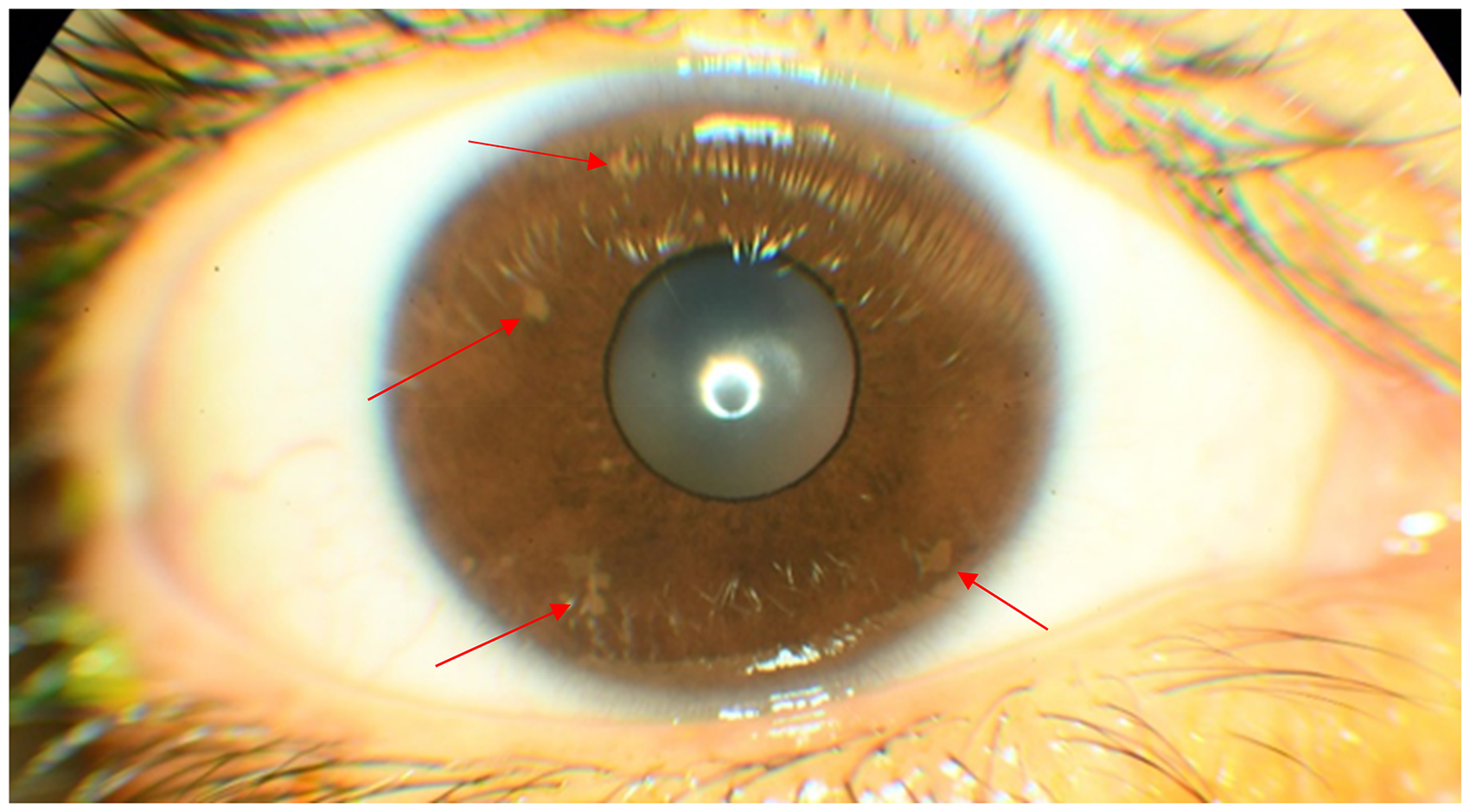
Lisch Nodules.
Optic glioma is a tumor of the optic nerve and is found in 15% to 20% of patients with neurofibromatosis type 1. It may be unilateral or bilateral. It is the most common cause of visual loss in neurofibromatosis type 1. Most gliomas associated with neurofibromatosis type 1 occur in the optic nerves, chiasm, optic tract, and optic radiations. Tumors grow slowly, have low-grade malignant potential, and are mostly pilocytic astrocytomas histologically. Clinically, neurofibromatosis type 1 may present with decreased visual acuity and color vision, abnormal pupillary function, optic atrophy, proptosis, nystagmus, and strabismus. Symptoms usually appear before the age of 6 years, and most children are diagnosed with optic glioma by the age of 3 years. Children with neurofibromatosis type 1 who show symptoms of optic tract glioma should undergo contrast-enhanced magnetic resonance imaging.3,6,8,13,17–21 Segal et al 17 found optic glioma in 44 (13%) of 331 paediatric patients (mean age 6 years) diagnosed with neurofibromatosis type 1. They reported that symptoms such as decreased vision or precocious puberty occurred in 8 of the patients. Isolated optic nerve involvement was observed in 43% of patients. 17 Optic gliomas do not require treatment if they are clinically stable. Chemotherapy is preferred for progressive optic gliomas. In this study, optic glioma was found with a rate of 7% (Figure 4).
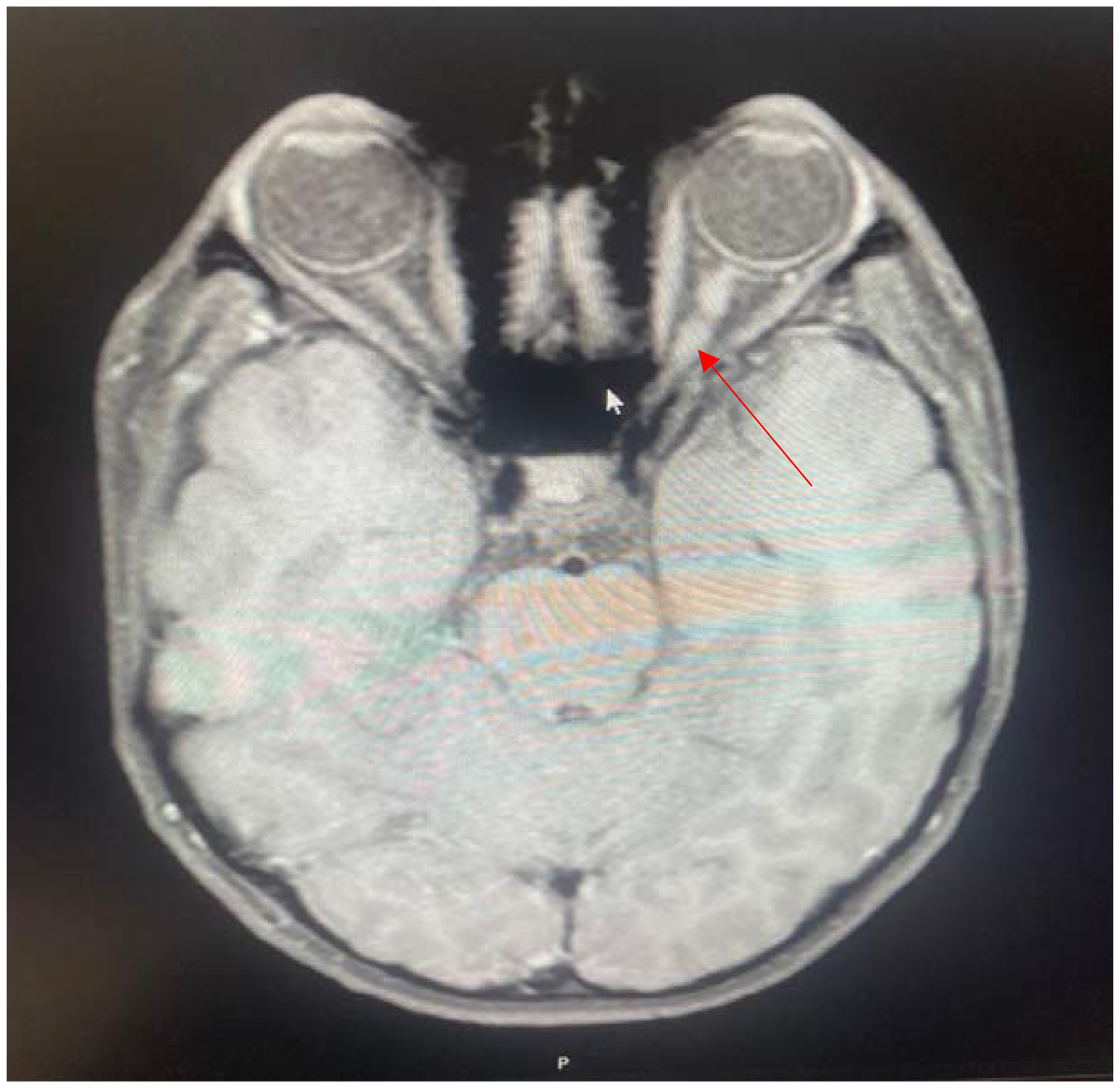
An Axial T1-Weighted Magnetic Resonance Imaging of Optic Glioma Showing Enlargement of the Left Optic Nerve.
Plexiform neurofibromas are complex tumors involving the nerves, connective tissue, muscle, vascular elements, and often overlying skin. These tumors can occur in various body parts; their occurrence in orbit is usually associated with sphenoid wing dysplasia. Eyelid plexiform neurofibromas cause thickening of the upper eyelid and S-shaped ptosis.3,7,8,14,21 Farris et al 7 reported in a study that all 10 patients presented with ptosis or lid swelling, and 2 patients presented with proptosis in addition to lid complaints. In this study, ptosis was recorded in 1 boy (1.4%) in the 0-6-year age group.
The findings of orbital neurofibromatosis include enophthalmos, exophthalmos, pulsatile exophthalmos, pulsatile enophthalmos, ptosis, facial-orbital deformity, and hypertelorism. Exophthalmos is frequently caused by an intraorbital mass formed by plexiform neurofibroma or glioma. Secondarily, it is caused by herniation of intracranial contents into the orbit as a result of congenital glaucoma or sphenoid wing dysplasia. Pulsatile exophthalmos occurs when intracranial pulsation passes through the bone defect into the orbit. Enophthalmos occurs because of the sagging of orbital contents into the infratemporal fossa as a result of the enlargement of the inferior orbital fissure.7,14,21 Neurofibromas that grow or have signs of malignant transformation and cause mechanical ptosis or cosmetic deformity are indicated for surgery. Complete excision is usually impossible because of the lesions’ infiltrative nature, and recurrence is quite common. Nerve preservation during surgery is challenging because neurofibromas are nonencapsulated tumors containing all nerve elements, axons, sheath cells, and connective tissues. Radiotherapy is avoided because of the danger of malignant transformation. 3
Congenital glaucoma is rare in neurofibromatosis type 1 and is usually unilateral. Glaucoma may also occur because of obstruction of humor aqueous flow by neurofibromatous tissue in the angle or closure of the angle as a result of neurofibromatous thickening of the ciliary body and forward displacement of the peripheral iris. In most cases, glaucoma is associated with plexiform neurofibromas on the same side. Medical and surgical treatment may be necessary to treat glaucoma.3,21
Retinal astrocytic hamartomas, retinal capillary haemangiomas, and retinal hamartomas are common retinal tumors in neurofibromatosis type 1. Retinal hamartomas sometimes cause retinal dialysis and tractional retinal detachment.3,5,21 Choroidal neurofibromatosis is characterized by oval bodies in the choroid. It is not possible to detect choroidal changes by simple fundus examination or fluorescein angiography. It is possible with indocyanine green angiography and near-infrared reflection imaging, a noninvasive tool showing choroidal neurofibromas as bright, patchy lesions.11,22
In prior studies, strabismus and refractive errors were found in children with neurofibromatosis type 1. Yossifon et al, 23 in a study performed in 76 patients, found strabismus in 22 patients (28.9%). It was reported that 5 patients (22.7%) had esotropia and 17 patients (77.2%) had exotropia. Dotan et al 24 stated that the incidence of myopia (28.5%), astigmatism (21.4%), and anisometropia (14.2%) was higher in children <12 years of age with neurofibromatosis type 1 disease than in the control group and frequently required optical correction. Akinci et al 25 found 23.1% myopia, 9.7% hypermetropia, and 19.5% astigmatism in their study performed in pediatric patients with neurofibromatosis type 1. Although the incidence of myopia was higher than the control group, there was no significant difference in the incidence of hypermetropia and astigmatism than in the control group. Kinori et al 21 recorded that the prevalence of mild to moderate myopia was higher in patients with neurofibromatosis type 1 than in the general population. In this study, strabismus was found in 5.6% of the 7-12-year age group, including 2 boys (2.8%) and 2 girls (2.8%). Myopia was found in 7.0%, hypermetropia in 4.2%, myopia astigmatism in 7.0%, hypermetropia astigmatism in 8.4%, and anisometropia in 7.0%. Dotan et al 24 reported in their study that the prevalence of astigmatism, a known risk factor for amblyopia, was higher in children with neurofibromatosis type 1. In our study, astigmatism was found to be more common than other refractive errors when myopic astigmatism and hyperopic astigmatism were evaluated together. Vision loss caused by refractive errors can be treated with optical correction, and early detection of refractive errors can prevent amblyopia.
Amblyopia is usually unilateral and is one of the most common causes of visual loss in children. It can be caused by any reason that creates a difference in vision between the 2 eyes. Mainly, strabismus arises from anisometropia. Amblyopia is observed in most children with orbitotemporal plexiform neurofibroma due to ptosis and anisometropia. 26 In this study, 5.6% had visual acuity <0.8 in the right and left eyes, 94.4% had visual acuity ≥0.8 in the right and left eyes, and 11.3% had amblyopia.
This retrospective study has limitations. The small number of patients and the fact that the patients’ information was limited to the recorded information limit this study. The strenghts of this study is that ocular findings associated with neurofibromatosis type 1 were analyzed in detail according to age groups, and the findings are consistent with the findings of previous studies.
Conclusions
Neurofibromatosis type 1 is a multisystemic disease, and ocular manifestations are common. Careful examination of the eyes enables the recognition of significant clinical signs of neurofibromatosis type 1 in childhood and the timely initiation of disease-specific treatment. Annual ophthalmologic examination of children with neurofibromatosis type 1 is crucial for improving the clinical course of the disease and preserving vision.
Footnotes
Ethical Approval
Ethical approval was obtained with the number 2024/6551 from the Ethics Committee of Inonu University Faculty of Medicine. This study was performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from all participants.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availibility
All data are available on formal request to the corresponding author.