
Abstract

Image Report
The ATP-binding cassette (ABC) superfamily encodes membrane proteins that carry various substrates along the membranes. These transporters are ATP-dependent pumps, and loss-of-function variations might lead to some different Mendelian disorders, including X-linked adrenoleukodystrophy (X-ALD) and cystic fibrosis. 1 X-ALD is a neurodegenerative disorder caused by pathogenic variations of the ABCD1 gene encoding a peroxisomal membrane protein, ALDP. Cystic fibrosis is a multisystemic disease resulting from biallelic CFTR gene variations encoding the ABCC7 transporter. Here, we describe a patient in which 2 or more variants in ABC protein genes were detected for, the first time in the literature; highlighting that atypical features may indicate a blended phenotype.
A 6-year-old male patient with attention-deficit hyperactivity disorder (ADHD) presented with sudden visual loss. Contrast-enhanced brain magnetic resonance imaging (MRI) findings were suggestive of X-linked adrenoleukodystrophy (Figure 1). He was found to have increased levels of very-long-chain amino acids as well (C26: 4.76 µmol/L [0.33-1.50 µmol/L], C24/C22: 1.76 µmol/L [0.44-0.97], C26/C22: 0.116 µmol/L [0.01-0.03]). His parents were nonconsanguineous and healthy. He had a brother, deceased due to X-linked adrenoleukodystrophy (Figure 2). The proband was shown to harbor a hemizygous missense variation in the ABCD1 gene (p.Pro560Leu, ENST00000218104.6, c.1679C > T, rs398123105) with Sanger sequencing analysis (Applied Biosystems, USA). It was a known “pathogenic” alteration, resulting in a shift in the primary structure of the protein. 2 It was confirmed the mother had this change, heterozygously. The proband's clinical condition deteriorated rapidly with loss of speech, muscle weakness, swallowing difficulties, respiratory distress, and eventually adrenal insufficiency. In the course of the disease, he also manifested intermittent episodes of pancreatitis, atelectasis in the respiratory tract, and Pseudomonas aeruginosa growth in bronchoalveolar lavage fluid culture. Targeted next-generation sequencing analysis for the CFTR gene showed that he had compound heterozygous variations. One of these (p.Val920Met, ENST00000003084.6, c.2758G > A, rs373885282) was a missense variant in a well-conserved residue. Its population frequency was low, and computational analysis predicted a mostly deleterious effect. As a result, it was interpreted as “probably pathogenic” according to the American College of Medical Genetics and Genomics guideline. 3 The other variant was c.1210-11T > G (rs73715573), a 5T;TG12 located in the polytract between the splice site in intron 9 and the acceptor site, predicted to affect mRNA splicing. It was reported as “pathogenic.” 4 Segregation analysis confirmed the CFTR variations were in the trans phase.
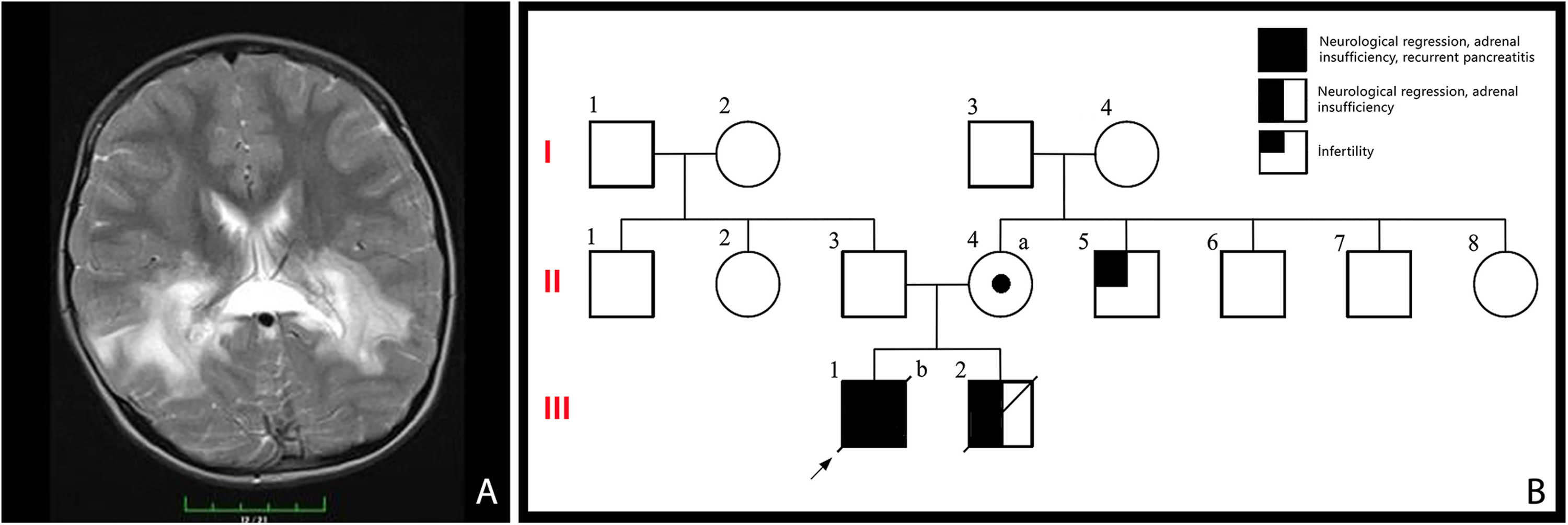
Clinical and genetic findings of the presented case. (A) In T2-weighted images, after intravenous gadolinium injection, intense subcortical contrast enhancements were observed in the splenium part of the corpus callosum and bilateral periatrial white matter areas and extending to the brainstem along the bilateral corticospinal tract. The measurements made from the left parietal area revealed an NAA/creatinine ratio of 0.57/1.1, which was significantly reduced, and a choline/creatinine ratio of 2.51/1.10, with an increase in this ratio, supporting the diagnosis of X-linked adrenoleukodystrophy (X-ALD). (B) Pedigree of the case, indicating II-4: A 31-year-old female with no health complaints, who was found to have a heterozygous P560L variation in the ABCD1 gene. II-5: A 28-year-old male with infertility who had not undergone any genetic testing, yet. III-1: The index case who had both adrenoleukodystrophy and pancreatitis. III-2: The case with hemizygous ABCD1 gene variation who was diagnosed with adrenoleukodystrophy; deceased at 3 years old after the stem cell transplantation. Informed consent for the publication of the patient's and their data were taken from the parents. NAA, N-acetylaspartate.
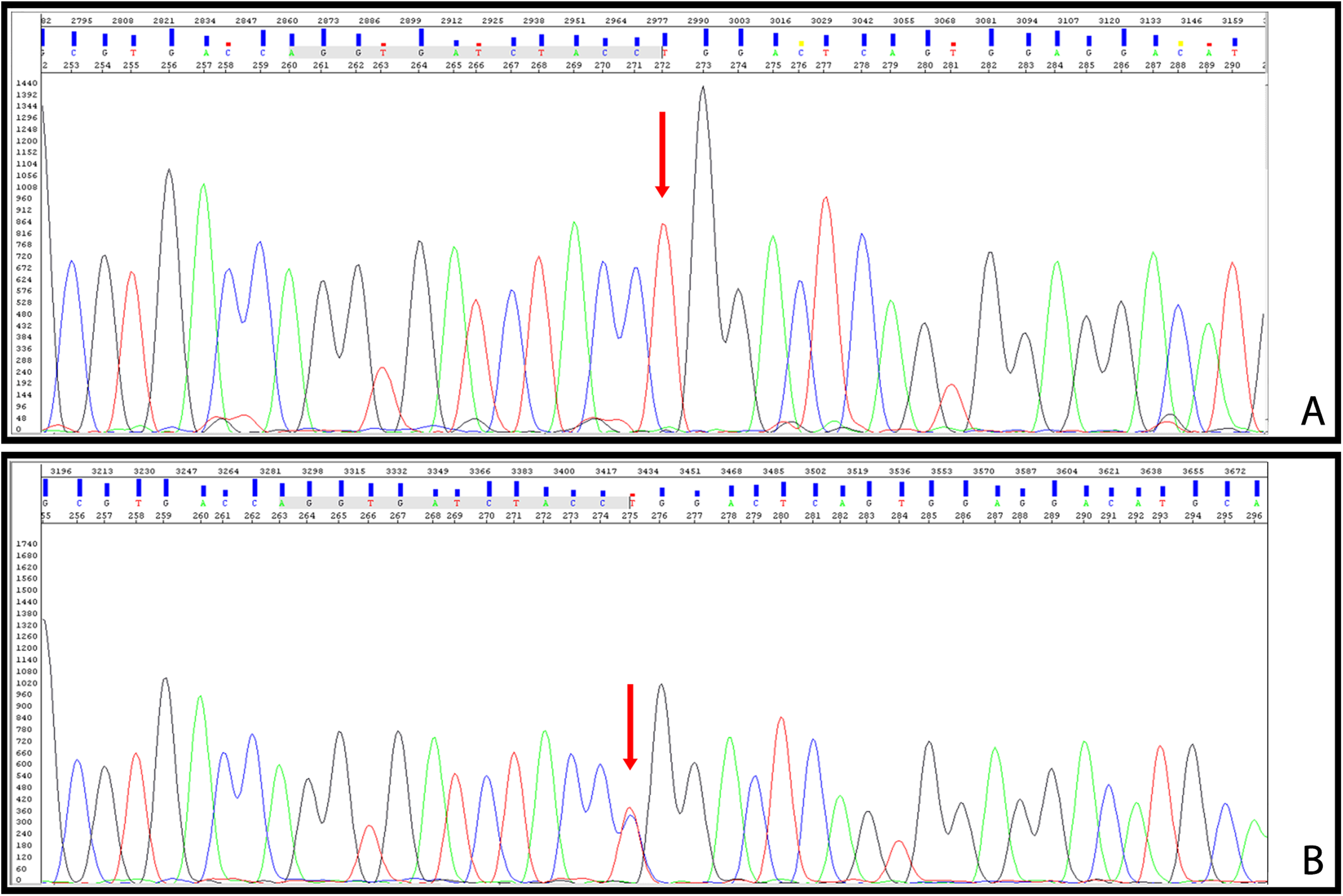
(A) Proband's Sanger sequencing plot for the chrX:153006072 region, with several base pairs surrounding it. Each peak represents a nucleotide read; the letter above each peak denotes the corresponding nucleotide identified at that position. The arrow indicates the hemizygous change (C>T) observed. (B) The mother's Sanger sequencing plot for the same region. The arrow indicates the location of the heterozygous change observed.
Our case was diagnosed with the cerebral phenotype of X-linked adrenoleukodystrophy with the first clinical findings of attention deficit and strabismus. Later he had epileptic seizures, neurologic regression, loss of vision, and muscle tone. Both symptoms and white matter demyelination were compatible with the diagnosis. 5 However, he had unexplained pancreatitis episodes and atelectasis which led us to another major diagnosis, cystic fibrosis. He was treated with hydrocortisone replacement for adrenal insufficiency; however, an episode of acute pancreatitis occurred during follow-up, which was managed conservatively. Coordination of care required a multidisciplinary team including pediatric neurology, endocrinology, and gastroenterology. At the age of 10 years, the patient died as a result of an adrenal crisis and accompanying pancreatitis. Following the dual diagnosis, the family received genetic counseling regarding the X-linked inheritance of ALD and autosomal recessive inheritance of cystic fibrosis. Carrier testing and extended family screening were recommended. The parents subsequently had a healthy child through assisted reproduction with preimplantation genetic diagnosis.
As a consequence, the coexistence of mutations in both ABCD1 and CFTR genes is presented to emphasize the mixed phenotype of a case. Further genetic testing might be needed for patients with unexplained symptoms, in order to achieve early and accurate diagnosis.
Footnotes
Author Contributions
KTÜ was the corresponding author of the article, the main designer of the manuscript, contributed to the acquisition of data, and drafted the initial version of the manuscript. BOA contributed to the conception, design of the study and writing the manuscript. BOA and AGZ made the genetic analyses. HÇ, SP, AY, and AGZ played important roles in the acquisition, analysis, and interpretation of the case data. HÇ, SP, AY, AGZ and BOA provided critical revision of the manuscript. All authors read and approved 4 1 the final version. All authors agree to be accountable for all aspects of the work in ensuring accuracy and integrity.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Ethical Approval
Ethical approval is not necessary for a case report at Necmettin Erbakan University Faculty of Medicine