
Abstract
Childhood disintegrative disorder is a poorly understood neurobehavioral disorder of early childhood characterized by acute to subacute profound regression in previously developed language, social behavior, and adaptive functions. The etiology of childhood disintegrative disorder remains unknown and treatment is focused on symptomatic management. Interest in neuroinflammatory mechanisms has grown with the increased recognition of autoimmune brain diseases and similarities between the presenting symptoms of childhood disintegrative disorder and pediatric autoimmune encephalitis. Importantly, a diagnosis of pediatric autoimmune encephalitis requires evidence of inflammation on paraclinical testing, which is absent in childhood disintegrative disorder. Here we report 5 children with childhood disintegrative disorder who were initially diagnosed with possible autoimmune encephalitis and treated with immunotherapy. Two children had provocative improvements, whereas 3 did not change significantly on immunotherapy. Additionally, a sixth patient with childhood disintegrative disorder evaluated in our Autoimmune Brain Disease Clinic showed spontaneous improvement and is included to highlight the variable natural history of childhood disintegrative disorder that may mimic treatment responsiveness.
Childhood disintegrative disorder is a rare but devastating neuropsychiatric disease first described in 1908 by Theodor Heller as “dementia infantalis.” 1 Childhood disintegrative disorder remains poorly understood, and though subsumed under autism spectrum disorder in the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5), 2 childhood disintegrative disorder remains separate in the International Classification of Diseases, Tenth Revision, reflecting the ongoing debate whether childhood disintegrative disorder is a separate process from autism spectrum disorder.3,4
Childhood disintegrative disorder has an estimated prevalence of 1 per 100 000, well over 100 times lower than autism spectrum disorder. 5 It is characterized by an acute to subacute onset of marked behavior changes, loss of language and adaptive skills, disinterest in the environment, and cognitive regression in children after more than 2 years of normal development.6–8 Children are typically 3-4 years of age at disease onset and follow either a static course or demonstrate partial improvement after the initial period of decline.6,7,9,10
Features distinguishing childhood disintegrative disorder from autism spectrum disorder include its notable rarity, longer periods of early normal development, faster initial course of regression, and more global developmental and symptomatic domains affected. Ultimately, children with childhood disintegrative disorder have lower IQ scores, more mental health symptoms, a higher incidence of epilepsy, and poorer general prognosis.6,11 Interestingly, the features that most distinguish childhood disintegrative disorder from autism spectrum disorder are similar to the presentation of children with certain forms of autoimmune encephalitis, which is also characterized by acute to subacute multidomain neurobehavioral regression with associated neuropsychiatric symptoms. Importantly, in addition to exclusion of other causes, a diagnosis of autoimmune encephalitis requires a positive paraclinical test demonstrating inflammation (ie, autoantibodies associated with autoimmune encephalitis, cerebrospinal fluid markers, magnetic resonance imaging [MRI], or brain biopsy findings),12,13 all of which are normal or not well reported in children with childhood disintegrative disorder.
As childhood disintegrative disorder is a clinical diagnosis that shares features with autoimmune encephalitis, consideration of childhood disintegrative disorder may be appropriate for some children with possible autoimmune encephalitis and vice versa.14,15 Accurate differentiation between these populations is crucial given the notable lack of specific treatments in childhood disintegrative disorder whereas immunomodulatory treatments may significantly impact the course of autoimmune encephalitis; however, misdiagnosis and treatment of patients without autoimmune encephalitis carry risk of immunomodulatory agent side effects and potential delay of optimal engagement in symptomatic management of childhood disintegrative disorder. 16 Often, when diagnostic testing is underway, a trial of immunomodulation is considered in patients with possible autoimmune encephalitis; however, current recommendations support that if no paraclinical marker of inflammation results, further treatment is not supported.16,17 In this context, we describe the clinical features and courses of 5 patients treated for possible seronegative autoimmune encephalitis with first- and second-line immunotherapy in whom a diagnosis of autoimmune encephalitis was ultimately not supported by updated criteria and replaced with a diagnosis of childhood disintegrative disorder. For comparison, a childhood disintegrative disorder patient who was not treated with immunotherapy is included to highlight the need for controlled studies and the importance of positive paraclinical markers of inflammation in the diagnosis of autoimmune encephalitis.13,16
Patients and Methods
Patient Selection
A single tertiary care center multidisciplinary pediatric autoimmune brain disease (ABD) clinic database was reviewed for patients with rapid-onset regression concerning for possible autoimmune encephalitis who were treated with immunomodulation for >6 months who ultimately did not meet criteria for probable or definite autoimmune encephalitis.
13
Cases were screened for those with documented normal development past 24 months of age with no significant premorbid neurologic or genetic diagnoses (eg, Autism, Trisomy 21, SHANK3)6,18 and a documented hyperacute (<1 week) or acute (<4 weeks) onset of multidomain profound neurobehavioral regression including at least 3 of the following:
Loss of language including inability to express needs and wants with minimal receptive language Loss of interest/awareness of surroundings or interactions with others and a loss of independent play Marked sleep disturbances with continuous periods of insomnia or excessive sleep Loss of basic age-appropriate adaptive functions such as feeding oneself, toileting, and/or dressing Marked agitation with periods of hyperkinetic behavior, excessive pacing and/or prolonged periods of persistent nonpurposeful movements or lack of movement (eg, catatonia-like symptoms)
These nonspecific neuropsychiatric symptoms seen in autoimmune encephalitis meet the DSM-IV childhood disintegrative disorder criterion
18
provided no other cause of symptoms were identified. An additional child evaluated in autoimmune brain disease clinic who met the above criteria but did not receive any immunomodulatory treatment was included for illustrative purposes. The Duke Institutional Review Board approved this retrospective case review study.
Chart Review and Symptom Scoring
Initial clinical and demographic data were collected from referral records. ABD physicians from pediatric neurology, rheumatology, and psychiatry evaluated all patients; detailed history was reviewed for premorbid development past 24 months of age. Subsequent clinical visits were reviewed with symptoms, examinations, testing, and treatments recorded.
Symptoms at the time of presentation, and subsequent clinical visits, were retrospectively scored by 3 reviewers with the Glasgow Outcome Scale–Extended. The Glasgow Outcome Scale–Extended was initially designed for use in patients with traumatic brain injury to define functional outcomes from death to good recovery on an 8-point scale and was used to provide an overall assessment of adaptive functioning. 19 Symptom burden using the Multidomain Assessment Scale for Autoimmune Encephalitis (MDA) was scored at initial presentation, time of referral to our multidisciplinary clinic, and at last evaluation. The MDA is a qualitative, provider-based symptom score assessing 6 functional domains commonly affected in autoimmune encephalitides. 20 Each domain is scored on an 8-point scale from most severe to normal. Lastly, the Severity Level of Autism Spectrum Disorders as defined by the DSM-V was used to characterize the residual phenotype given the prominent autism spectrum disorder features seen in chronic phase childhood disintegrative disorder.2,6 Discrepancies in scoring were resolved through discussion.
Results
Patient Characteristics and Testing
Demographics, clinical history, and genetic testing are presented in Table 1. All patients were male with an average age at onset of 5.5 years (range 3.9-8.3, SD = 1.48). Clinical concern and diagnostic testing for metabolic, degenerative, infectious, and genetic causes varied widely. Subsequent anti–streptolysin O and anti–DNase B antibody titers were normal in both patients with prodromal positive streptococcal antigen testing. Serum and cerebrospinal fluid testing for infectious etiologies were uniformly negative (eg, viral studies and mycoplasma antibodies). Genetic testing was performed in 5 of 6 patients, with analyses varying over time and between insurances. No results had previously been associated with regression or childhood disintegrative disorder specifically; however, 2 of 5 chromosome microarray analyses showed copy number variants associated with autism spectrum disorder. Both the 15q11.2 microdeletion (patient 4) and 1q21.2q21.2 duplication (patient 5) were paternally inherited without neurodevelopmental symptoms in their fathers. Expanded testing was completed in 4 patients with no pathogenic mutations identified (Table 1).
Demographics, Clinical History, and Genetic Evaluation.
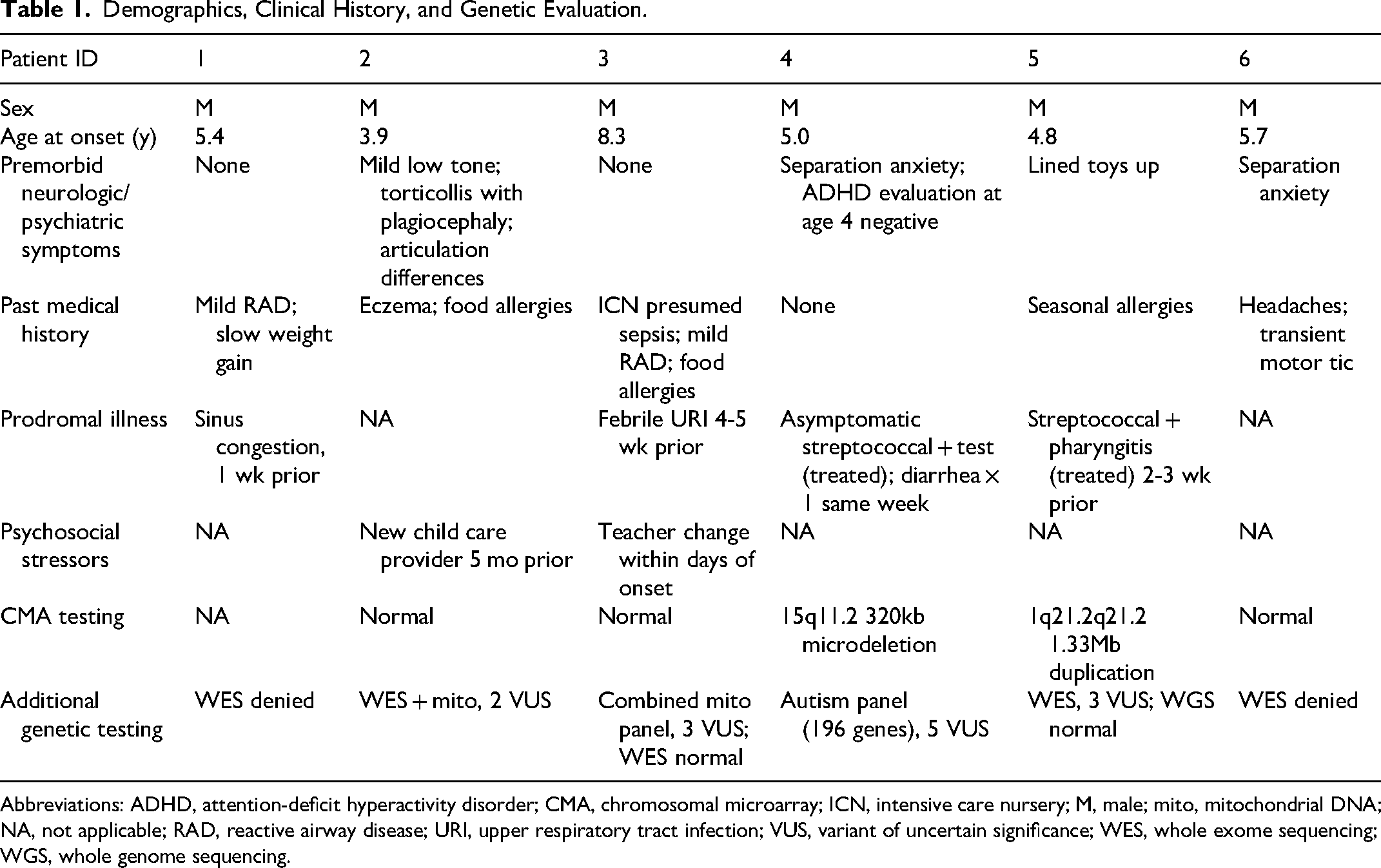
Abbreviations: ADHD, attention-deficit hyperactivity disorder; CMA, chromosomal microarray; ICN, intensive care nursery; M, male; mito, mitochondrial DNA; NA, not applicable; RAD, reactive airway disease; URI, upper respiratory tract infection; VUS, variant of uncertain significance; WES, whole exome sequencing; WGS, whole genome sequencing.
All children had uniform evaluations for autoimmune encephalitis without paraclinical evidence of inflammation (Table 2), including normal brain magnetic resonance imaging (MRI) with gadolinium, serum markers of inflammation (erythrocyte sedimentation rate and C-reactive protein), serum and cerebrospinal fluid autoimmune encephalitis antibody panels; cerebrospinal fluid testing showed no pleocytosis, or elevations in protein, IgG indices, or oligoclonal banding. One patient had a borderline elevation in thyroid peroxidase antibodies; all others were negative at presentation. Two patients had nondiagnostic brain positron emission tomography (PET) scans. Initial electroencephalograms (EEGs) were abnormal in 3 of 6 and in an additional patient on repeat evaluation. No patient had definite seizures, although staring episodes and incontinence were common.
Diagnostic Evaluation for Autoimmune Encephalitis.a
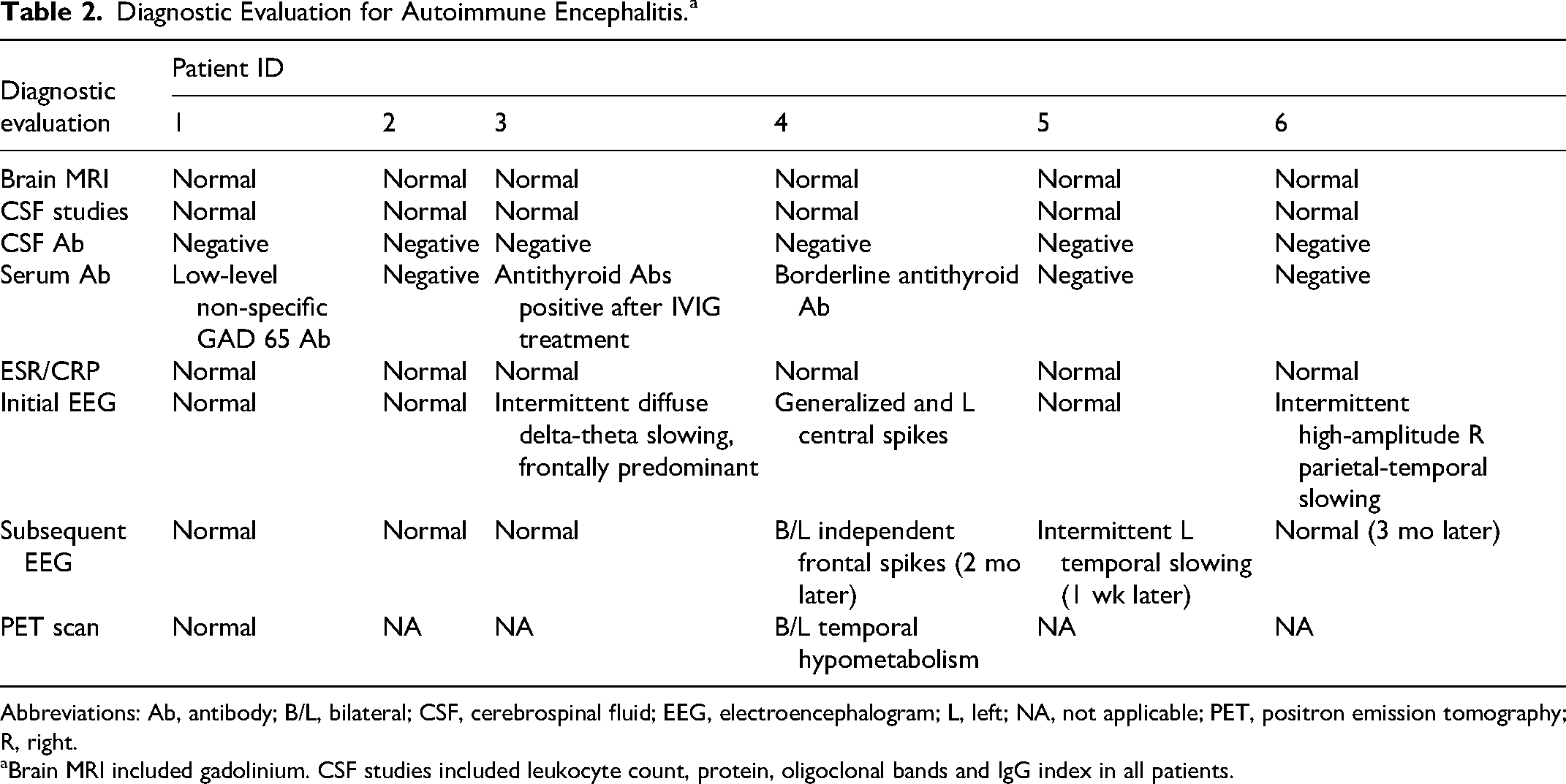
Abbreviations: Ab, antibody; B/L, bilateral; CSF, cerebrospinal fluid; EEG, electroencephalogram; L, left; NA, not applicable; PET, positron emission tomography; R, right.
Brain MRI included gadolinium. CSF studies included leukocyte count, protein, oligoclonal bands and IgG index in all patients.
Presenting Symptoms and Disease Courses
Clinical symptoms are presented in Table 3. All patients experienced profound language impairment (5 of 6 mute), cognitive regression, social impairment, loss environmental awareness and play behavior. Incontinence occurred in 5 of 6 and marked insomnia in 3 of 6. All patients displayed agitation (prominent pacing in 4 of 6), hand stereotypies, decline in fine motor skills, tics, obsessive compulsive disorder, and psychosis (including 3 of 6 with Capgras delusions).
Clinical Symptomatology.
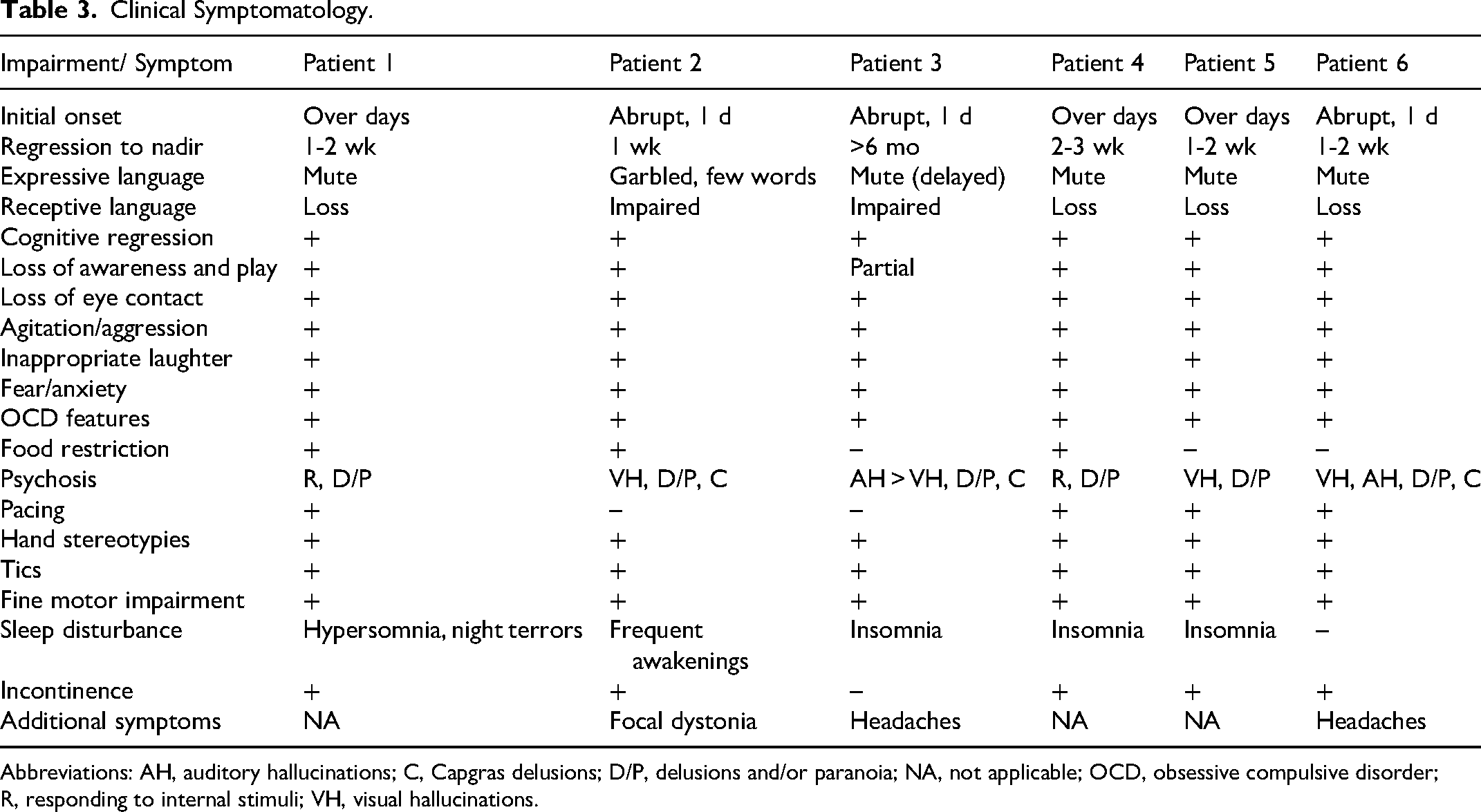
Abbreviations: AH, auditory hallucinations; C, Capgras delusions; D/P, delusions and/or paranoia; NA, not applicable; OCD, obsessive compulsive disorder; R, responding to internal stimuli; VH, visual hallucinations.
Patients were referred to our clinic an average of 9 months after onset (range 2.5-19.3 months) and after treatment with immunotherapy was started in each case. Longitudinal changes in adaptive functioning based on Glasgow Outcome Scale–Extended scores and concurrent immunomodulatory treatment exposure are shown in Figure 1. Two patients (patients 1 and 2) had early treatment with initial partial improvement in adaptive function and language noted on intravenous immunoglobulins (IVIGs) that plateaued, followed by further gains with escalation of immunotherapy. These patients are considered possible partial responders. Two additional patients (patients 4 and 5) also had early immunotherapy but with modest improvement in play and adaptive function, with minimal improvement in language, cognitive, social, and behavioral domains despite escalations. Patient 3 had a longer interval after onset of psychosis before cognitive regression developed and immunotherapy was started. After treatment, he had mildly fluctuating subjective symptom reports with no objective improvements seen and even some further progression despite escalation; he is considered a nonresponder. Lastly, patient 6 highlights the spontaneous improvement that may occur in childhood disintegrative disorder; this improvement was similar to that in patients 1 and 2 and would likely have been misinterpreted as a response to immunomodulation had it been started in the interval.
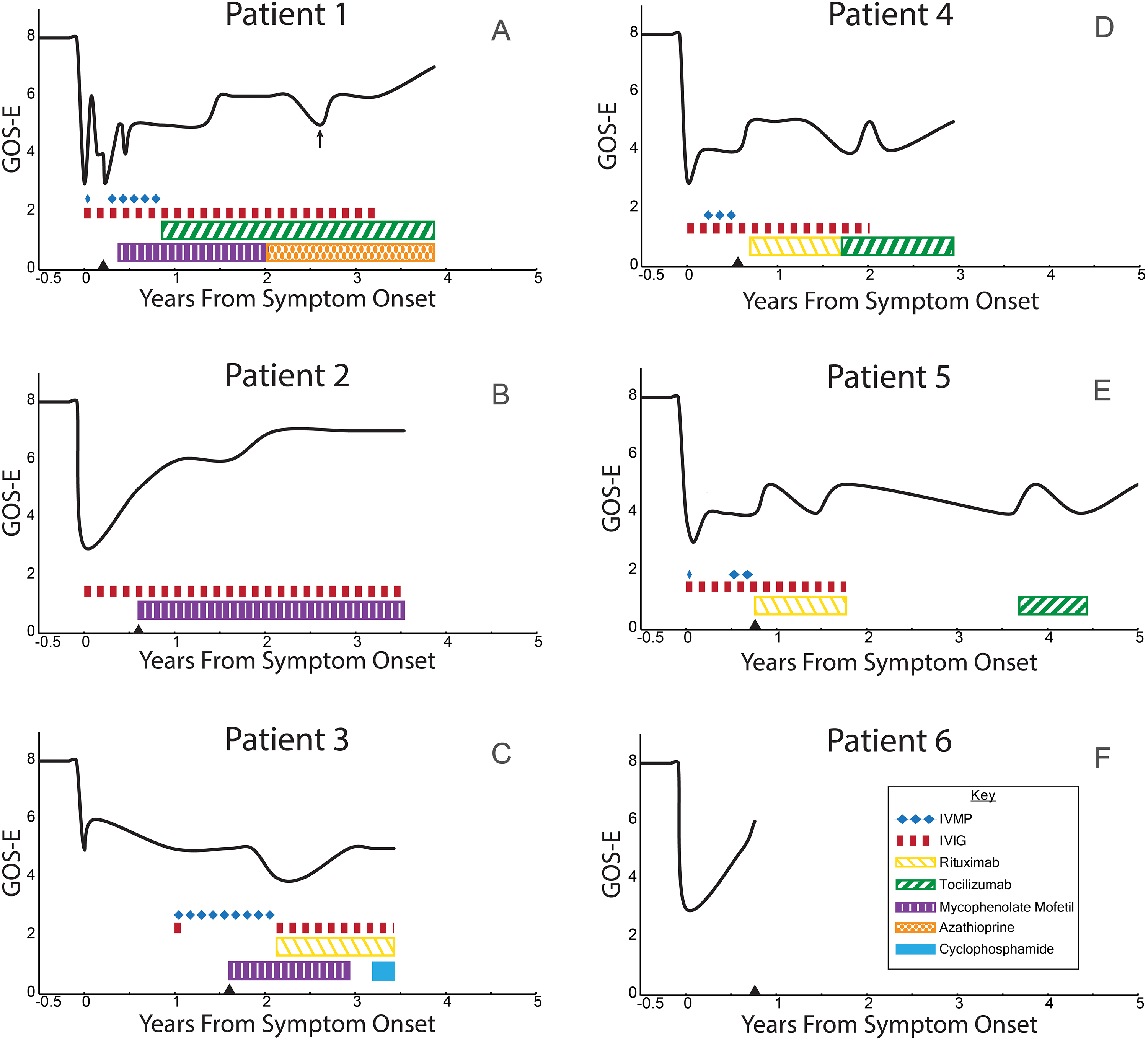
Disease courses and immunotherapy. Adaptive function scores from the Glasgow Outcome Scale–Extended are shown over time for patients 1-6 in panels A-F, respectively. Immunotherapy agent and duration are as shown with IVIG and IVMP (IV methylprednisolone) given as pulse doses every 3-6 weeks (dashes do not reflect specific doses). Triangle denotes time of autoimmune clinic referral. Arrow (patient 1) shows a pseudo-flare related to anxiety exacerbation.
Symptom scores from MDA categories (cognition, language, psychiatric, and sleep) are shown in Figure 2 at onset, referral to our center, and at last ABD clinic follow-up (average 3.3 years, range 0.8-5 years). Psychosis improved early in 5 of 6 but was replaced by anxiety, obsessive compulsive disorder, and autism spectrum disorder features within the psychiatric symptom domain. Agitated catatonia improved early but attention-deficit hyperactivity disorder (ADHD) / impulsivity continued. Sleep improved, but often with notable pharmacologic management in place. All patients improved in terms of fine motor control, orientation, environmental awareness, play behavior, cooperation with assisted activities of daily living, word utterances, and receptive language skills; however, only cases 1, 2, and 6 had more functional language, ADL independence, and learning improvements seen (Figure 2).
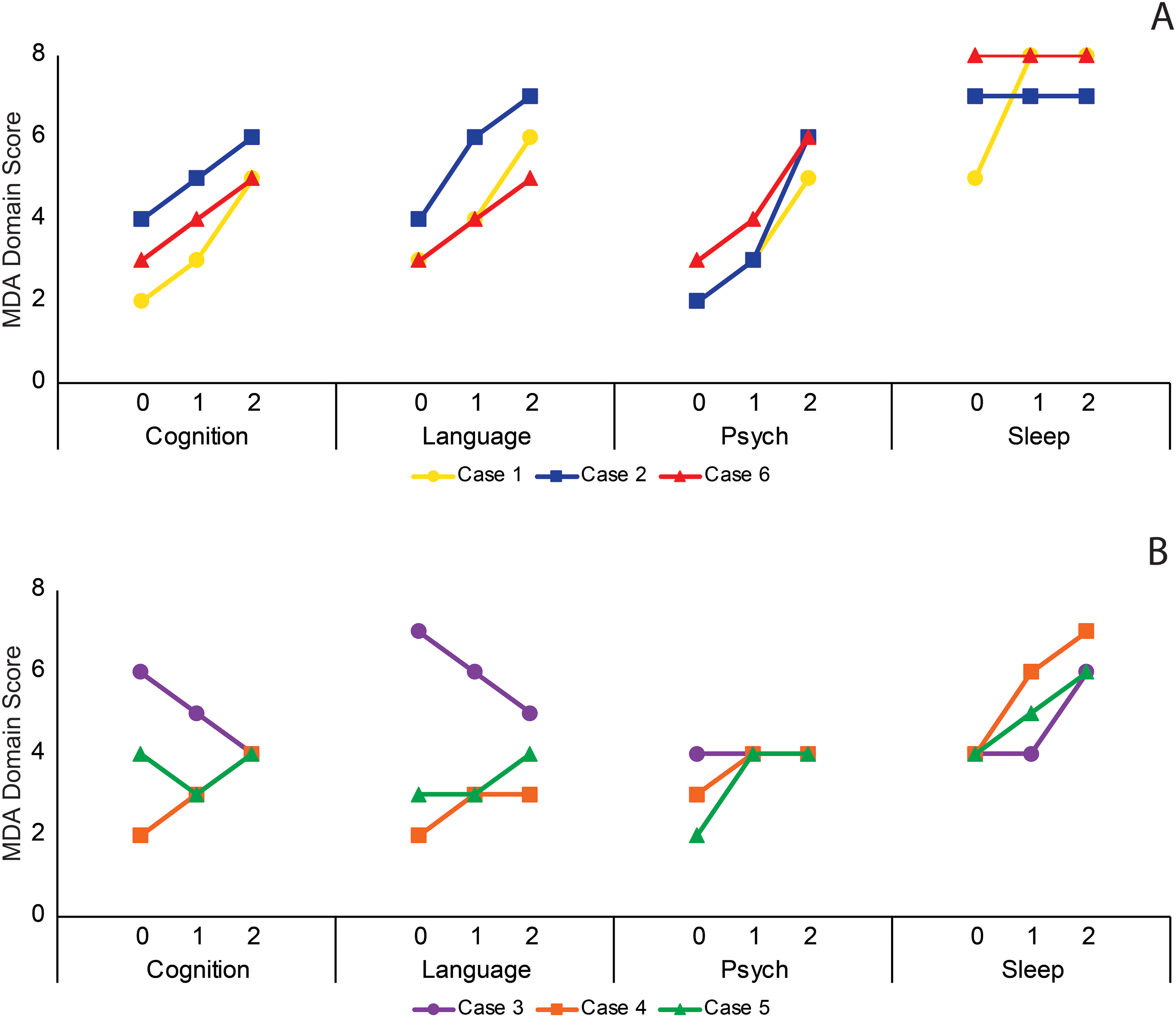
Symptom domain changes by outcome. Patients are grouped by overall outcome as those having (A) mild residual disease or (B) moderate to severe residual disease. Symptom domain scores from the MDA are graphed over 3 time points: (0) presentation, (1) referral to our clinic, and (2) last evaluation. Seizure and Motor domains are not included.
Case Vignettes
Patient 1
This normally developing 5-year-old male presented following sinus congestion with acute onset of new food contamination fears, eisoptrophobia, lack of play, increased sleep, repetitive hand movements, and pacing. He developed mutism, poor memory, anorexia, incontinence, decreased eye contact, limpness, and hallucinations. Workup was unremarkable. Methylprednisolone did not improve symptoms and he was treated with IVIG. Within a few days, he was speaking in phrases, eating, and less agitated with no signs of psychosis. Five weeks later, he declined with repeat IVIG, again correlating with improvement. Six weeks later, notable regression recurred over a week and repeat workup remained unremarkable, including PET scan. IVIG (1 g/kg) was scheduled monthly and lorazepam was started for agitated catatonia. His psychotic and catatonia-like symptoms improved readily with only modest improvements in language, social, play, and adaptive skills over several months; he was successfully weaned off lorazepam. His symptoms plateaued and mycophenolate mofetil (MMF) was added without benefit followed by tocilizumab with improvement noted in receptive and expressive language, memory, social interest, oral intake, sleep, and participation in assisted activities of daily living. Mycophenolate mofetil was switched to azathioprine. More than 2 years after onset, quetiapine and fluoxetine improved pseudo-regression symptoms secondary to heightened anxiety and ADHD behaviors coincident to school reentry (arrow, Figure 1). He remains in partial school days in a regular classroom with special services for academics and therapy services. His psychotic and catatonic behaviors remain in remission; his language, cognitive skills, and adaptive function are improved but notably impaired, with neuropsychometric testing showing most domains ≤1st percentile. In addition, he has generalized anxiety, tics, and obsessive compulsive disorder features that fluctuate in severity (with intercurrent illnesses and stressors) and a functional autism spectrum disorder level of 1. IVIG and tocilizumab are being weaned (developed hyperlipidemia); he remains on azathioprine, quetiapine, and fluoxetine. His course and response to immunotherapy was provocative and considered to be a possible partial response.
Patient 2
This 4-year-old man had low tone with torticollis and plagiocephaly treated as an infant and some separation anxiety and articulation differences as a toddler but normal core developmental milestone acquisition. Five months prior to presentation, he had difficulty sleeping and tantrums after changing childcare providers. These had improved prior to him becoming abruptly confused at preschool. He did not recognize his parents and was screaming in fear with Alice in Wonderland syndrome perceptions. Hospital evaluation was unrevealing, including normal cerebrospinal fluid opening pressure. He was disoriented with garbled speech and third person references to self, hallucinations, contamination fears, sensory sensitivities, incontinence, emotional lability, impaired memory, and further language regression with inability to follow commands or perform ADLs. He was noted to have new intermittent right leg dystonia and right hand chorea. He was treated with 2 g/kg IVIG every 3 weeks with improvement in psychosis, language, and ADLs but continued agitation, confusion, incontinence, obsessive compulsive disorder, and anxiety. After IVIG, he was noted to have papilledema and repeat lumbar puncture showing elevated opening pressure of 34 mm H2O, treated with acetazolamide. After partial improvement with IVIG plateaued, he was started on mycophenolate mofetil and guanfacine with school accommodations and intensive therapy services in place. He had further improvements in overall functioning, including improved behavioral, anxiety, and obsessive compulsive disorder symptoms. Escitalopram was briefly trialed for anxiety. He is in school with an aide and therapy services; he continues to have occasional right leg dystonia and a level 1 autism spectrum disorder phenotype. Mycophenolate mofetil is slowly weaning, and he remains on monthly IVIG as well as hydroxyzine and guanfacine for symptomatic management. His course and response to immunotherapy was provocative and considered to be a possible partial response.
Patient 3
This previously normally developing 8-year-old boy had a febrile URI ∼1 month before he abruptly became withdrawn and fearful, reporting he was hearing voices. He developed insomnia, vocal and motor tics, fine motor decline, disruptive and aggressive behavior, and his grades declined. Brain MRI was normal. His psychosis progressed with 2 psychiatric hospital admissions including treatments with clonidine, olanzapine, fluvoxamine, and valproate with partial improvement in hallucinations. He developed a sinus infection and worsened anxiety, tics, and obsessive compulsive disorder symptoms. One year after his initial decline, autoimmune encephalitis evaluation showed only an abnormal EEG background. His behavior and paranoia worsened and methylprednisolone (monthly) and IVIG (2 doses) were started for possible autoimmune encephalitis. His hallucinations, interactions, and tics improved but frequent headaches, intermittent unintelligible speech, anxiety, obsessive compulsive disorder, and insomnia continued. He was started on mycophenolate mofetil with some improvement in anxiety and behavior reported. At 2 years from onset, he continued to deteriorate cognitively, became mute, and was no longer making any eye contact. He was reinitiated on IVIG (notable nausea, vomiting, and headaches) followed by rituximab with subjective improvements in paranoia, hallucinations, and sleep reported. Numerous psychotropic medications were trialed with modest results. He continues to require full supervision and assistance in ADLs with severe Level 3 autism spectrum disorder. After returning to local providers, he underwent a course of plasmapheresis and remains unchanged clinically. His course is different than the others with an older age of onset, more persistent psychosis and delayed cognitive regression over many months; childhood-onset schizophrenia could be considered as an alternate diagnosis to childhood disintegrative disorder. He had no discernable response to immunotherapy.
Patient 4
This patient was described as shy, but without an anxiety diagnosis who underwent evaluation for ADHD at age 4 years that was negative. At age 5 years, he experienced abrupt changes in behavior including inappropriate laughter, emotional lability, impulsivity, hyperactivity, social withdrawal, and incontinence. He had one bout of diarrhea and an asymptomatic pharyngeal streptococcal antigen test was positive; he was treated with antibiotics without benefit. He continued to decline, developing mutism, stereotypies, pacing, restrictive eating, hypersexual behavior, insomnia, obsessive compulsive disorder, aggression, and hallucinations. At evaluation, EEG showed generalized and left central spikes and minimal elevation in thyroid peroxidase antibodies. He was started on lamotrigine followed by oxcarbazepine empirically (no seizures). He received methylprednisolone (caused agitation) and IVIG. Symptomatic treatment of insomnia included clonidine, melatonin, and diphenhydramine. There was minimal change in his status, and he was retreated with methylprednisolone and IVIG. Some improvements in sleep and aggression were reported and he said a word or two. He continued on IVIG monthly; sleep and psychotic symptoms improved without language, cognitive, or behavioral changes. Rituximab was added and he was reported as calmer; applied behavioral analysis and intensive therapies were put in place. PET scan showed nonspecific mild bilateral medial temporal hypometabolism. A subsequent trial of tocilizumab had no effect. He developed acquired hypogammaglobulinemia and remains on monthly replacement IVIG. Over 3 years after his initial presentation, he says a few words, has some echolalia, follows some commands, plays with preferred toys; he has continued aggression, ADHD, obsessive compulsive disorder, poor language skills, poor adaptive functioning, and a level 3 autism spectrum disorder phenotype. He attends school in a contained classroom with multiple aides and therapy services. His course is considered improved from his nadir without clear response to immunotherapy.
Patient 5
Developmental history showed normal milestones and play but noted he liked to “line up” toys. Three weeks prior to presentation at 4.8 years, he had streptococcal pharyngitis treated with antibiotics. He then acutely developed anxiety, visual hallucinations, Capgras delusions, insomnia, receptive and expressive language loss, and staring episodes. Initial workup was normal; subsequent EEG showed intermittent left temporal slowing. He had impairing anxiety, hypersensitivity to clothes and baths, tics, inappropriate laughing, writhing hand movements, pacing, social impairment, and incontinence. Methylprednisolone worsened symptoms. Oxcarbazepine, clonidine, clonazepam, zolpidem, doxepin, and risperidone were trialed without improvement. About 3 months after presentation, he had some return of language but was babbling and intermittently catatonic. IVIG and methylprednisolone were scheduled monthly with slow minimal improvements in receptive language and modest interaction with family that waxed and waned. He demonstrated some play behaviors but with severe anxiety, poor adaptive functioning, and hypersexual behavior. Rituximab was started with minimal changes noted. His hallucinations returned, and a throat swab tested positive for streptococcal antigen, treated with antibiotics. Over time, he returned to school in a contained classroom, he regained continence, cooperated better with assisted ADLs, played independently with restricted interests, and would speak in short phrases rarely, but with ongoing insomnia, anxiety, obsessive compulsive disorder, ADHD, and aggression. Later in his course, he was trialed on tocilizumab without additional benefit. He continues to have a marked level 3 autism spectrum disorder phenotype. His course is considered improved from his nadir without a clear response to immunotherapy.
Patient 6
Patient was a normally developing 6-year-old with separation anxiety as a toddler, a history of headaches, and transient motor tic of childhood. One day in virtual school, he abruptly froze, was unresponsive, and had urinary incontinence. Over the next weeks he had rapid regression in language and cognition with inappropriate laughter and mood lability. He developed hallucinations, confusion, aggression, staring spells, pacing, tics, hand fidgeting, incontinence, and needed full assistance with ADLs. Workup revealed intermittent focal slowing on EEG but was otherwise unremarkable. He plateaued after the initial several weeks of decline. About 5 months after presentation, he began to have spontaneous improvement without having received any immunomodulatory or symptomatic treatments. He started at a new school and was more social again, language started to return, pacing reduced, and hallucinations stopped; he regained toileting skills and needed less help with ADLs. Less than 1 year after onset, he had spontaneous speech, could answer most questions, follow directions, and was starting to relearn lost cognitive abilities. Our follow-up interval was very short in his case as he was not being treated with immunotherapy. Residually, he has a mild level 1 autism spectrum disorder phenotype. His spontaneous improvement >5 months after onset was similar in clinical pattern to that of patients 1 and 2.
Discussion
Here we describe the presentation and course of 5 children with abrupt onset neurobehavioral regression with psychosis treated with immunomodulation for possible autoimmune encephalitis, and an additional patient who did not receive immunotherapy. Paraclinical testing was uniformly normal, with none meeting updated criteria for probable or definite autoimmune encephalitis 13 ; furthermore, all have a residual autism spectrum disorder phenotype and meet published criteria for diagnosis of childhood disintegrative disorder.2,18 Our cohort experienced typical core symptoms consistent with published reports of childhood disintegrative disorder.6,18,21 Although subtypes of childhood disintegrative disorder may have an acute or more insidious onset, our inclusion criteria required acute to subacute onset, similar to the presentation of autoimmune encephalitis with profound impairments and psychiatric symptoms, likely contributing to referral bias. Our cohort also had prominent agitated catatonia and pacing, hand stereotypies, staring episodes, inappropriate laughter, poor eye contact, and incontinence. Likely reflecting referral patterns for possible autoimmune encephalitis, all 6 reported here had psychosis compared to 33% in a recent meta-analysis; conversely, no patient in this cohort had definite seizure activity compared with 36% in reports (although 4 of 6 had an abnormal EEG at some point). 6
Autistic regression and psychosis as seen in this cohort have been identified as potential presentations of autoimmune encephalitis.12–14,22 Thus, etiologic evaluation for autoimmune encephalitis should be considered in addition to possible genetic, metabolic, infectious, and trauma-reactivity causes of rapid-onset neurobehavioral regression. Of note, EEG background changes are not paraclinical markers of inflammation and are common in both autism spectrum disorder and childhood disintegrative disorder. 6 Depending on clinical suspicion, providers may start immunotherapy while awaiting testing results. Ideally, if no evidence of inflammation is noted, treatment would stop and alternate diagnoses sought. Previously, a clinical response to immunomodulation was a criterion for diagnosis of autoimmune encephalitis. 23 However, the nonspecific effects of first-line immunotherapy agents and challenges related to subjective symptom reporting have deemphasized “response to treatment” as a criterion for diagnosis; the importance of paraclinical markers of inflammation for accurate autoimmune encephalitis diagnosis has also been demonstrated.13,16,24 In this context, it is notable that 2 of the patients presented here appeared to have building responses to escalating immunotherapy after a plateau in symptoms on IVIG alone. Each, however, also received substantial school and therapy supports, symptomatic medical management, and have a notable residual cognitive/behavioral phenotype despite treatment. Furthermore, the contrast of case 6 highlights that in addition to contributions from symptomatic interventions, delayed spontaneous improvement may be falsely attributed to concurrent immunotherapy. Importantly, nearly all our patients had resolution of early psychosis, improved environmental awareness and regained continence despite notable residua consistent with outcomes reported in childhood disintegrative disorder that include partial improvement after initial decline.6,7 In the chronic phase, our cohort continues to have residual symptom fluctuations and pseudo-flares with stressors and illnesses that may also be misinterpreted as related to weaning of immunotherapy. Despite these confounding factors, the disease courses in patients 1 and 2 are provocative and warrant consideration of well-designed, controlled trials for selected patients given the often devastating outcomes of childhood disintegrative disorder. This must be balanced with the risks of chronic immunotherapy including infection, infusion-related side effects, metabolic derangements, and acquired hypogammaglobulinemia as seen in several patients here.
Treatment of childhood disintegrative disorder remains symptomatic with intensive behavioral and educational interventions and psychopharmacotherapy targeting mood, agitation, sleep, anxiety, obsessive compulsive disorder, and ADHD symptoms. Proposed etiologies for childhood disintegrative disorder include primary psychiatric, genetic, epileptic, immune/inflammatory, and aberrant neurocircuitry. Behavioral or primary psychiatric etiologies were previously suggested by frequent prodromal stressors and the prominent behavioral dysregulation during the initial decline.1,9,25 Disease manifestation during periods of acute psychological (or physiological stress), are non-specific however. 26
Support for a genetic basis for childhood disintegrative disorder has been noted in reports of sibling clusters,25,27 with associations to copy number variants, and in candidate gene clusters.4,28–30 In our cohort, chromosome microarray revealed a 15q11.2 BP1-BP2 microdeletion in patient 4 causing Burnside-Butler syndrome. These patients show developmental and speech disorders in 67% to 73% and autism spectrum disorder, ADHD and behavioral problems in 25%-55%, with another 20% having schizophrenia. 31 The 1q21.1q21.2 1.33Mb microduplication found in Case 5 is associated with ADHD and autism spectrum disorder in 30%-40% of patients and has cognitive and expressive language deficits commonly noted.32,33 Both copy number variants were paternally inherited with known incomplete penetrance and variable expressivity. As childhood disintegrative disorder is notably more rare than autism spectrum disorder, copy number variants associated with autism spectrum disorder are likely relevant in this cohort despite not previously being documented with childhood disintegrative disorder explicitly. Reported VUS genes were all associated with developmental delays, often autism spectrum disorder, but not regression explicitly. Increasing data regarding the degree and nature of genetic abnormalities related to autism spectrum disorder / childhood disintegrative disorder and the variability in testing methods used here make the role of genetic changes in our cohort unclear.
An epileptic etiology has also been suggested in childhood disintegrative disorder given the relatively high incidence of seizures and abnormal EEGs in this population6,7,25 and the similar regressive phenotypes in children with continuous spike waves during slow-wave sleep (CSWS) and related epileptic aphasias. 34 The etiology of these epileptic encephalopathies is varied with symptomatic, cryptogenic, and genetic causes known. Clinical response to corticosteroids and other immunomodulating agents are reported,35,36 but improvement with benzodiazepines and antiseizure medications is also seen. 37 Based on the age of onset and Hebbian mechanisms of synaptic strengthening, it has been theorized that abnormal circuitry and deficient pruning results in regional patterns of disrupted neural network development leading to long-term developmental sequela, even after the EEG abnormalities and seizures subside. 34 Interestingly, some of the genetic links found in CSWS patients are also reported in autism spectrum disorder and schizophrenia populations.31,38 Studies of cytokine/chemokine levels in CSWS showed differences from controls that returned toward normal after immunotherapy, correlating to improved EEG and behavior, 39 but patients treated successfully with antiseizure medications were not included, limiting interpretation of this result.
Recently, autism spectrum disorder research has increasingly focused on broad immune/inflammatory biomarkers. Clinically, Mordekar et al reported successful corticosteroid treatment in 2 children with a childhood disintegrative disorder phenotype; however, both had prominent high voltage slowing on EEG suggestive of an epileptic encephalopathy 40 instead of the milder EEG features commonly reported in childhood disintegrative disorder. 7 More abundant pharmacotherapy data in autism spectrum disorder and that with regression 41 include >18 “immune pathway” drug studies, including corticosteroids and IVIG.42–44 Small sample sizes, phenotypic and biochemical heterogeneity, and most important, subjective parent-reported outcomes greatly limit interpretation of these overwhelmingly positive results. Interestingly, IVIG was one of the few agents often showing no benefit. 42 Putative targets have been actively sought in studies designed to elucidate possible immune or inflammatory derangements in autism spectrum disorder, with components of both the innate and adaptive immune systems often showing differences, peripherally and within the central nervous system (eg, increased pro-inflammatory cytokines/chemokines, autoantibodies to brain antigens, altered complement profiles, and activation of microglia).45–47 Microglial activation and complement protein changes are often thought to be inflammatory in this context; however, microglia and the complement system subserve cellular support, neural network development, synaptogenesis, and dendritic pruning as well.48,49 Alterations in these functions may link to an alternate (noninflammatory) theory that autism spectrum disorder is a disorder of aberrant neural connectivity.
The concept of altered neural connectivity and deficient pruning has been considered historically with possible underlying mechanisms including aberrant electrical synaptic strengthening, 34 deficient IGF-1 cascade cellular signaling, 50 and disrupted oligodendrocyte function and myelination. 51 Disconnectivity is hypothesized to create an excitation/inhibition imbalance that may lead to oxidative stress and subsequent inflammation.50,51 In autism spectrum disorder, as with many conditions, it is challenging to determine whether inflammation is causal or in consequence to primary pathologies; if in consequence, inflammation may further contribute to disruption propagation or its reversal.
This is the first report of sustained immunotherapy in children with childhood disintegrative disorder. Our results demonstrate 2 of 5 treated children with a provocative response, although this should be interpreted with caution as effects may be overestimated based on variability in the natural history of childhood disintegrative disorder and concomitant symptomatic treatments in our patients. Referral to our center was likely influenced by the same criteria we used for inclusion (abrupt onset, severity of regression) but also potentially biased by additional factors, such as prominence of psychotic symptoms and parental behavior. All had initial treatment started locally, contributing to variable time to treatment with variability in immunotherapy based on patient course, occasional co-management with outside providers, mechanism of action considerations, and changes in practice over time, further limiting interpretation of treatment response. The workup for autoimmune encephalitis was uniform, but those for metabolic, infectious, and genetic testing varied. Additional limitations include small sample size, retrospective design, and subjective symptom reports to assess adaptive functioning (Glasgow Outcome Scale–Extended) and symptom burden Multidomain Assessment Scale for Autoimmune Encephalitis (MDT), neither of which have been validated in children with autoimmune encephalitis or childhood disintegrative disorder.
Conclusions
Childhood disintegrative disorder is a poorly understood and notably devastating disease in which previously typically developed children suffer an often rapid and severe loss of language, adaptive function, and cognition that subsequently stabilizes to emerge with a severe autistic phenotype in most cases. After the initial regression, some children experience a mild to moderate improvement in symptoms resulting in less severe outcomes, raising important questions about potential reversibility of the initial pathophysiology. Prompt diagnostic evaluation for potentially treatable causes, including autoimmune encephalitis, should be strongly considered in children with a childhood disintegrative disorder phenotype, especially those with acute onset. However, treatment with traditional immunotherapy in patients with childhood disintegrative disorder is not currently supported by the literature on autoimmune encephalitis or autism spectrum disorder. Expert consensus recommendations for the overall diagnostic evaluation of children with childhood disintegrative disorder are needed clinically to identify mimickers, whereas more studies investigating the interplay between candidate pathologies, including aberrant neural network formation, synaptic pruning, reaction to oxidative stress, and inflammatory responses are needed to elucidate potential treatment targets.
Footnotes
Author Contributions
Author contributions included a substantial contribution to the concept or design of the work (CP, KT, HVM); or acquisition, analysis or interpretation of data (MS, MG, KT); drafted the article (MG, MS, CP) or revised it critically for important intellectual content (CP), approval of the version to be published (MS, MG, KT, HVM, CP); and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved (MS, MG, KT, HVM, CP).
Author Note
The views expressed in the submitted article are not an official position of Duke University Medical Center.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
The Duke University Medical Center Institutional Review Board approved this retrospective case review study.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.