
Abstract
Severe fever with thrombocytopenia syndrome (SFTS) is an emerging zoonotic disease caused by the tick-borne SFTS virus (SFTSV), with a case fatality rate of 16.2–32.6% in East Asia. Currently, no approved vaccines or antiviral treatments exist. Monoclonal antibody (mAb) therapy offers rapid immune protection and is a promising strategy against SFTSV. This review outlines advances in SFTSV neutralizing mAb research, covering conventional generation methods (hybridoma, phage display) and innovative approaches such as single B cell sequencing. We also introduce computational tools like artificial intelligence -assisted epitope prediction and in silico mAb design. Furthermore, we discuss the structure–function relationships of mAbs targeting Gn and Gc glycoproteins, their mechanisms (e.g., fusion inhibition, receptor blockade), and key functional attributes including breadth, potency, and cross-reactivity. Challenges such as limited epitope accessibility, immune interference, and antibody-dependent enhancement are highlighted. Finally, we propose a multipronged strategy integrating structure-guided engineering, high-throughput screening, and rigorous preclinical evaluation to accelerate the development of safe and effective SFTSV therapeutics.
Background
Severe fever with thrombocytopenia syndrome (SFTS) is a tick-borne hemorrhagic fever caused by SFTS virus (SFTSV) (Yu et al., 2011). Between 2011 and 2021, a total of 18,902 confirmed cases of SFTS were reported in China, resulting in 966 cases of deaths. The average annual incidence was 0.125 per 100,000, with a case fatality rate of 5.11%. Notably, the incidence exhibited a significant upward trend (Cui et al., 2024). The majority of affected patients are elderly individuals (Ding et al., 2014). Confirmed cases of SFTSV infection have been reported in Asian countries (China, South Korea, Japan, Vietnam, Myanmar, Thailand, and Pakistan) since 2010 (Li et al., 2022), indicating the expanding geographical scope of SFTSV. The primary route of SFTSV infection is tick transmission, with Haemaphysalis longicornis acting as the principal host and vector (Luo et al., 2015). Current epidemiological evidence suggests that domestic animals, such as dogs and cats, may serve as reservoirs for SFTSV and facilitate its transmission to human (Huang et al., 2019). SFTS is notable for its unique capacity for person-to-person transmission via blood contact, a feature not commonly observed in most other endemic zoonotic diseases (Gai et al., 2012). Clinical manifestations of SFTS are diverse, typically including high fever, thrombocytopenia, and leukopenia. In the early stages, patients often present with nonspecific symptoms such as fatigue and myalgia. These symptoms may progress to severe multi-organ dysfunction, potentially leading to death (Li et al., 2018a). Patients with severe SFTS show reduced Immunoglobulin G (IgG) and IgM levels in early infection compared with mild cases. Fatal cases often lack Gn-specific IgG and NP-specific IgM and IgG antibodies (Chung et al., 2022; Song et al., 2018). This deficiency may result from SFTSV-induced disruption of high-affinity antibody maturation and plasma cell differentiation, leading to impaired neutralizing antibody production, increased viral replication, and higher mortality (Suzuki et al., 2020).
SFTSV is officially named Dabie bandavirus and classified in the genus Bandavirus of Phenuiviridae family, Bunyavirales order (ICTV, 2023). As with other Bunyaviridae members, SFTSV has a tripartite negative-sense RNA genome, consisting of the S, M, and L segments. The L segment encodes RNA-dependent RNA polymerase, which is essential for transcription and replication of viral genome (Wang et al., 2020a). The M segment encodes the envelope glycoproteins Gn and Gc, derived from proteolytic cleavage of a glycoprotein precursor, which are essential for viral entry. Gn consists of a head domain and a stem region, with the head domain forming a compact triangular structure composed of three subdomains (I, II, and III). The stem region contains 10 conserved cysteines, four of which mediate disulfide-linked dimerization, a conserved feature among bunyaviruses (Du et al., 2023). Cryo-EM analyses indicate that Gn anchors to viral membrane via its stem region, while its head domain engages host receptors, such as Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin (DC-SIGN), NMMHC-IIA, and CCR2 (Hofmann et al., 2013; Sun et al., 2014; Tani et al., 2016; Zhang et al., 2023). Structurally homologous to class II fusion proteins, Gc contains a central fusion loop and a C-terminal transmembrane domain. In the prefusion state, Gc exists as a head-to-tail homodimer, resembling flavivirus E proteins. Upon endosomal acidification, it undergoes conformational rearrangement into a postfusion homotrimer, driving viral-endosomal membrane fusion. Notably, the metastable Gc fusion loop is shielded by Gn in the prefusion state, preventing premature activation (Yuan and Zheng, 2017). Ultimately, Gn and Gc form heterodimers that assemble into pentameric and hexameric spike structures on the virion surface (Du et al., 2023). The S segment encodes nuclear protein (NP) and nonstructural protein (NSs). NP is the most abundant protein in both SFTSV particles and infected host cells and plays a pivotal role in viral replication and assembly (Lee et al., 2023). NSs play a critical role in immune suppression by inhibiting the type I interferon (IFN-α/β) response, a key antiviral signaling pathway (Khalil et al., 2021). NSs suppresses type I IFN expression by inhibiting the IFN-β promoter and interacts with TBK1, disrupting Interferon Regulatory Factor (IRF) and NF-κB signaling pathways. This weakens antiviral responses and enhances SFTSV replication in monocytes, facilitating sustained viral replication and spread (Qu et al., 2012).
Despite the rising incidence and high mortality of SFTS, no approved vaccines or specific antiviral therapies are currently available (Zhang et al., 2022). Favipiravir (T-705), a broad-spectrum RNA polymerase inhibitor, has demonstrated antiviral efficacy but is associated with adverse effects, including hyperuricemia, elevated bilirubin, hepatotoxicity, and gastrointestinal disorders (Li et al., 2021c). Similarly, baloxavir, an endonuclease inhibitor, shows promise against SFTSV but may cause diarrhea, headache, and delirium (Wang et al., 2020b). Neutralizing monoclonal antibodies (mAbs) offer rapid and effective protection against viral infections. Advances in mAb discovery platforms—such as hybridoma technology, phage display, and single B cell sequencing—have enabled the identification of broadly neutralizing mAbs against SFTSV. This review summarizes current progress and future directions in SFTSV neutralizing mAb research. We first outline mAb generation strategies, from conventional methods to newer approaches like single B cell sequencing, and their roles in SFTSV mAb development. Emerging computational tools, including artificial intelligence (AI)-assisted epitope prediction and in silico mAb design, are also discussed for their potential to accelerate discovery. Next, we examine structure–function relationships of reported SFSTV neutralizing mAbs, focusing on their epitope specificity for Gn and Gc glycoproteins, mechanisms such as fusion inhibition and receptor blockade, and key functional properties including breadth, potency, and cross-reactivity. Finally, we highlight current challenges—such as limited epitope accessibility, immune interference, and the risk of antibody-dependent enhancement (ADE)—and propose a structure-guided, multipronged strategy combining high-throughput screening and rigorous preclinical evaluation to advance the development of effective SFTSV mAb therapeutics.
Developmental strategies for MAbs against SFTSV
Hybridoma technology
Hybridoma technology enables continuous production of mAbs by fusing antigen-immunized B cells with immortal myeloma cells, generating stable hybridomas (Köhler and Milstein, 1975) (Fig. 1). This method preserves natural heavy and light chain pairing and in vivo affinity maturation (Yin et al., 2024), ensuring sustained production of high-affinity, antigen-specific mAbs (Zaroff and Tan, 2019). These properties make it a valuable tool for developing SFTSV-targeted therapeutics and diagnostics.
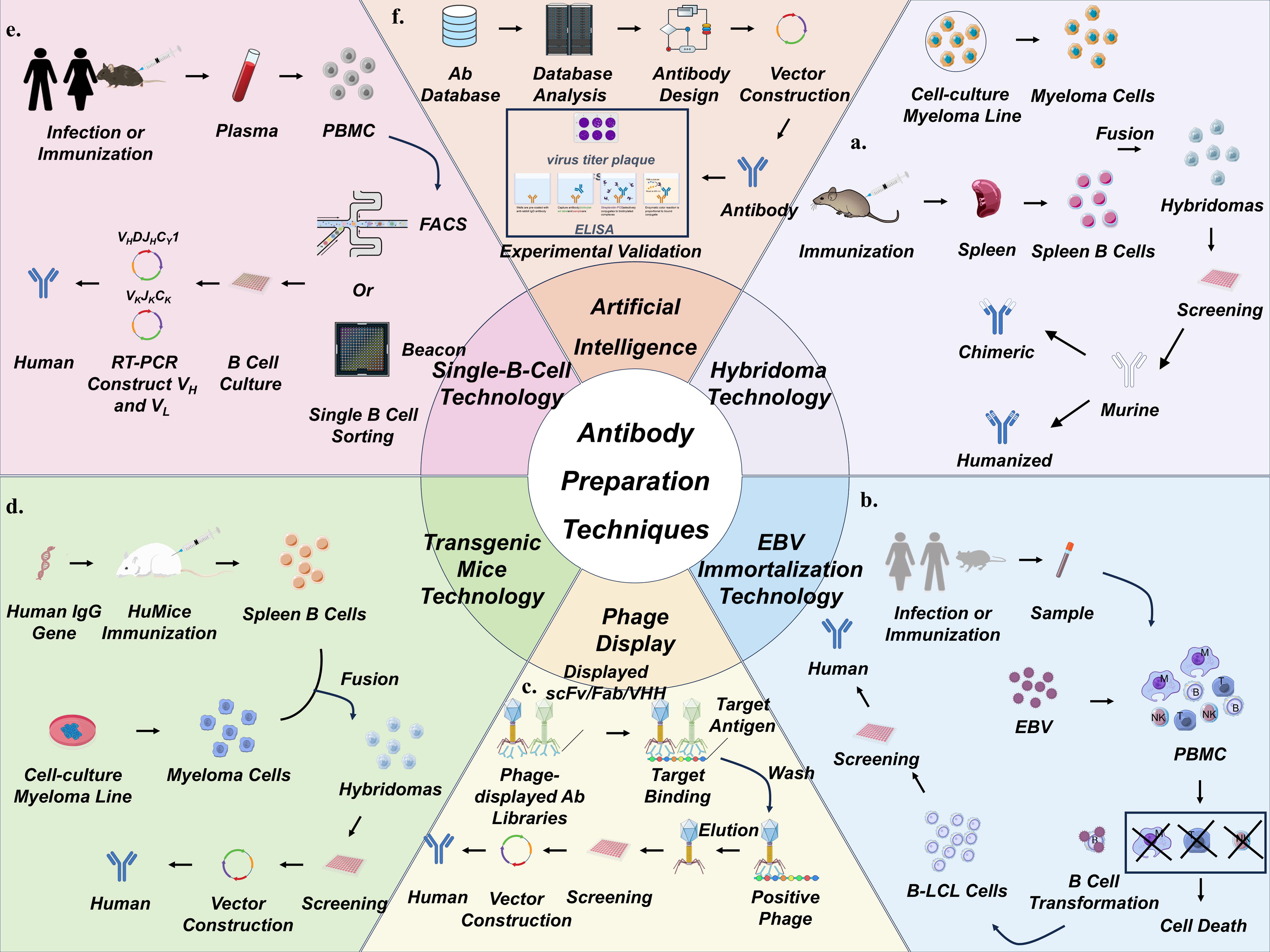
Strategies for the preparation of therapeutic mAb.
Hybridoma technology has enabled the development of SFTSV-specific mAbs with high sensitivity. Fukuma et al. successfully generated mAbs 9D3 and 2D11 targeting NP in BALB/c mice (BALB) (Fukuma et al., 2016), achieving detection limits as low as 10 pg/100 μL by sandwich Enzyme-Linked Immunosorbent Assay (ELISA). Since NP is not present on the viral surface, these antibodies are suited for diagnostics rather than neutralization. Hybridoma-derived mAbs C3A11, C6C1, and C4H12 target the Gc fusion loop (e.g., Trp652, Phe699), potentially stabilizing its prefusion state and blocking pH-dependent membrane fusion (Sano et al., 2024). Synergistic neutralization was observed when C6C1 or C4H12 was combined with the anti-Gn mAb MAb4-5, enhancing potency up to eightfold. No synergy was seen with mAb Ab10, likely due to epitope overlap causing steric or functional interference (Halldorsson et al., 2016). Despite strong in vitro activity, these mAbs showed limited in vivo efficacy in IFNAR−/− mice, possibly due to factors like epitope escape, short half-life, or poor tissue distribution. These findings highlight the need for in vivo evaluation of antibody combinations and support the development of bispecific or multi-epitope strategies for improved therapeutic outcomes. Wu et al. (2024) developed the broadly neutralizing anti-Gn mAb 40C10 using hybridoma technology. This mAb targets a conserved epitope in domain I of the SFTSV Gn protein, shared across genotypes C1–C4 and related viruses such as Heartland virus and Guertu virus. Structural studies revealed that 40C10 neutralizes via multiple mechanisms, including viral stabilization, particle aggregation, and fusion inhibition (Fig. 2), rather than solely blocking receptor attachment. To reduce potential HAMA responses, Yang et al. (2024) humanized mAb 40C10 through Complementarity-Determining Region (CDR) grafting, generating mAb HAb-23. Structural optimization and bioinformatics-guided evaluation preserved affinity while minimizing immunogenicity. HAb-23 showed potent neutralization (IC50 = 17.36 ng/mL) and provided both therapeutic and prophylactic protection in mice. Further studies are needed to assess its immunogenicity and define dosing for clinical use.
Schematic illustration of the mechanisms of SFTSV-neutralizing antibodies. Left panel: SFTSV infection process in host cells.
Murine mAbs often trigger HAMA responses and show limited ability to engage the human complement system. Due to poor interaction between murine Fc regions and human Fc receptors, these mAbs are less effective in mediating Antibody-Dependent Cell-Mediated Cytotoxicity (ADCC) and CDC (Hosono et al., 1992), reducing their clinical efficacy.
Phage display
Phage display enables the presentation of antibody fragments such as single-chain variable fragment (scFv), antigen-binding fragment (Fab), and nanobody (VHH) on the phage surface by inserting their genes into the phage genome (Lee et al., 2007) (Fig. 1). This allows high-affinity mAbs to be selected through antigen-binding screens. Unlike hybridoma technology, phage display is animal-independent and offers greater antibody diversity, making it a key tool in antibody engineering (Schirrmann et al., 2011).
Jeon et al. (Jeon et al., 2024) used phage display to identify 10 high-affinity Fabs against the SFTSV Gc protein, which were humanized into IgG1 mAbs. These mAbs showed strong binding to Gc with KD values as low as 15 pM (ELISA) and were validated by Surface Plasmon Resonance. Immunofluorescence assays confirmed broad cross-reactivity with SFTSV-infected cells from multiple genotypes (A, B2, B3, D, F), supporting their potential for serological diagnosis and Gc-targeted therapeutic mAb development. Guo et al. (2013) identified mAb MAb4-5 from a phage display library derived from lymphocytes of convalescent SFTS patients. MAb4-5 (Table 1) neutralized SFTSV clinical isolates from Jiangsu, Anhui, and Shandong, though its efficacy was slightly reduced (80–90%) against some Jiangsu strains (e.g., JS-2011-004, JS-2012-020). Western blot confirmed binding to the Gn extracellular domain (aa20–452). Structural analysis later showed MAb4-5 targets the α6 helix in subdomain III of Gn, an internal epitope likely obscured by Gn/Gc heterodimers (Wu et al., 2017). As such, MAb4-5 may act by disrupting glycoprotein assembly rather than preventing receptor binding. While broadly neutralizing against Chinese isolates, its efficacy against strains from other regions (e.g., Korea, Japan) and in vivo protection remain limited due to epitope inaccessibility. Kim et al. (Kim et al., 2019) isolated the mAb Ab10 (Table 1) from a phage-displayed scFv library using peripheral blood mononuclear cells (PBMCs) from a convalescent SFTS patient in Korea. Ab10 recognizes a conformational epitope spanning domain II and the stem region of the Gn glycoprotein, with key residues K371, R324, and C356. It likely blocks low pH-induced exposure of the Gc fusion loop, preventing membrane fusion (Fig. 2). In vitro, Ab10 potently neutralized SFTSV in Vero cells, reducing infection from 90% to 5.6% at 50 μg/mL, outperforming MAb4-5. It bound with high affinity (KD = 104 pM) to Gn proteins from diverse strains (Gangwon/Korea/2012, HB28, SD4), potentially covering 90.8% of global isolates. In A129 mice, a single 30 mg/kg dose provided full protection against lethal challenge, with 80% survival even when treatment began five days postinfection.
Summary of SFTSV Antibodies
SFTSV, severe fever with thrombocytopenia syndrome virus.
However, Ab10’s efficacy has not been validated in nonhuman primates, and detailed structural data are lacking. Its modest protective dose and limited neutralization data warrant further investigation. Wu et al. (2020) constructed a phage-displayed nanobody library from immunized camels and identified SNB02 (Table 1) as a potent antiviral candidate. SNB02 inhibited SFTSV infection in Vero E6 cells and human PBMCs (IC50 ≤ 2.7 μg/mL) and reduced viral load by >2 log10 in a humanized NCG Human Peripheral Blood Lymphocyte (NCG-HuPBL) mouse model when administered one-hour post-infection. It also alleviated thrombocytopenia, vascular leakage, and inflammatory infiltration in key organs. SNB02 likely blocks viral entry by targeting the Gn glycoprotein and interfering with receptor binding (Fig. 2). It recognizes a conserved epitope across SFTSV subtypes A–E and demonstrates broad cross-reactivity. Its small size (12–15 kDa) allows for deep tissue penetration and access to hidden epitopes. SNB02 remains stable without cold-chain storage and shows low immunogenicity due to its camelid Variable domain of Heavy chain of Heavy-chain antibody (VHH) scaffold, removing the need for humanization. However, its short half-life (∼9.5 h) in mice requires frequent dosing. Strategies like Fc fusion or PEGylation may extend circulation time. In addition, since the NCG-HuPBL model lacks key SFTS symptoms, further validation in nonhuman primates is needed.
Phage display has advanced SFTSV mAb discovery but faces key limitations. First, some antibody fragments may misfold or fail to display properly. Second, the absence of posttranslational modifications like glycosylation limits selection against glycan-dependent epitopes (Newby et al., 2024). Third, antigens are often immobilized in non-native conformations, masking transient or cryptic sites such as the Gc fusion loop (Du et al., 2023). Immunodominant decoy antigens, particularly the highly immunogenic NP, can skew phage libraries toward non-neutralizing antibodies (Li et al., 2018b; Wang et al., 2019; Yu et al., 2012), hindering the identification of rare neutralizing antibodies targeting Gn or Gc. To overcome these issues, strategies such as using pseudoviruses or native-like glycoprotein complexes can better present conformational epitopes. In addition, rational antigen design and optimized immunization protocols can reduce decoy dominance and enhance the yield of broadly neutralizing candidates.
Single B cell sequencing
Single B cell sequencing enables efficient mAb discovery by isolating antigen-specific B cells, amplifying IgG variable region genes, and expressing mAbs in mammalian cells (Pedrioli and Oxenius, 2021). Sorting methods include magnetic-activated cell sorting (Polyakova et al., 2023), fluorescence-activated cell sorting (FACS) (An and Chen, 2018), and microfluidics (Mehling and Tay, 2014). The Beacon® optofluidic platform (Fig. 1) offers high-throughput screening and precise cell handling (Winters et al., 2019). Compared with hybridoma methods, it preserves native heavy/light chain pairing, improving affinity and screening efficiency.
Single B cell technology has advanced SFTSV mAb discovery. Chang et al. (2024) identified two mAbs, SF5 and SF83 (Table 1), from convalescent individuals. SF5 targets domain I of Gn, recognizing buried residues (e.g., A85, E151, F83) potentially involved in receptor binding. Despite moderate neutralization, SF5 has low affinity due to limited somatic hypermutation. SF83 binds the inner face of domain II on Gc (K710), disrupting Gc assembly. While SF83 shows strong in vitro binding, its in vivo efficacy is limited, likely due to poor epitope accessibility. To enhance efficacy, a bispecific mAb, bsAb3, was engineered by combining SF5 and SF83 in a DVD-Ig format. bsAb3 sequentially targets Gn and Gc during viral entry, blocking membrane fusion (Fig. 2). It showed potent neutralization in vitro (low IC50) and full protection in vivo at 5 mg/kg, while limiting viral escape. However, findings are limited to the A129 mouse model. Further evaluation in nonhuman primates and assessment of manufacturing scalability and clinical stability are needed. Zhang et al. (2025) used single B cell technology to isolate broadly neutralizing mAbs from four convalescent SFTS patients. JK-2 and JK-8 (Table 1) showed potent neutralization (FRNT50 < 100 ng/mL) against five genetically distinct SFTSV strains, including Chinese lineages I–IV and Japanese lineage K-13. Both mAbs improved survival in mice when given prophylactically or up to three days post-infection. Structural studies revealed that JK-2 and JK-8 target domain I of Gn. JK-8 forms stable interactions via hydrogen bonds and salt bridges, including contacts with the N63 glycan, blocking Gn-CCR2 binding and preventing viral entry (Fig. 2). Their epitopes are more exposed than those of Ab10 or MAb4-5, allowing efficient engagement on both pentameric and hexameric virion surfaces. However, the study was limited by the small donor pool and restricted viral lineage exposure. Pharmacokinetic properties also remain uncharacterized. Ren et al. (2024) generated the mAb S2A5 (Table 1) using single B cell sequencing after mouse immunization. S2A5 showed broad neutralization against six SFTSV genotypes (A–F) and provided both prophylactic and therapeutic protection in mice with a single dose. Mechanistically, S2A5 targets domain I of the Gn glycoprotein, blocking viral attachment and fusion. It interacts with 17 key residues and crosslinks adjacent Gn protomers via its light chain, enhancing neutralization. Compared with other mAbs, S2A5 offers strong potency, broad genotype coverage, and dual inhibitory mechanisms. However, its epitope lies near domain II of Gn, potentially causing steric interference. Genotype-dependent variation in pseudovirus neutralization was noted but not fully validated in authentic strains. In vivo protection has only been tested against a genotype D strain, which is uncommon in China and South Korea.
Despite its advantages, single B cell technology faces limitations in SFTSV mAb discovery. A key challenge is the reliance on recombinant Gn and Gc antigens, which may not fully mimic native conformations. Misfolded or nonphysiological antigens can hinder the isolation of B cells targeting cryptic or pH-dependent epitopes. To overcome this, recent efforts have focused on stabilizing viral glycoproteins to preserve native structures, enabling more accurate retrieval of functional, physiologically relevant mAbs.
Artificial intelligence
AI has shown great promise in accelerating the design and screening of mAbs against SFTSV (Fig. 1). High-performance models like RFdiffusion (Watson et al., 2023) and AlphaFold-Multimer (Omidi et al., 2024), which predict protein–protein interactions, can be fine-tuned with curated structural data, including SFTSV antigens and epitopes, to improve accuracy. RFdiffusion can optimize mAbs targeting specific epitopes on Gn and Gc proteins, while ProteinMPNN (Dauparas et al., 2022) refines CDR loop sequences. Incorporating SFTSV-specific datasets into models like RoseTTAFold2 (Humphreys et al., 2024) further enhances their ability to distinguish true antibody–antigen pairs from decoys. This approach improves the efficiency of candidate screening before experimental validation.
AI has also become a valuable tool for analyzing B cell receptor (BCR) repertoires from convalescent individuals to identify broadly neutralizing mAbs. High-throughput sequencing generates extensive BCR heavy- and light-chain sequences, which AI analyzes using tools like Abalign (Zong et al., 2023) to perform clonotype clustering, V(D)J gene assignment, and mutation analysis. This enables rapid identification of high-frequency clones associated with neutralizing activity against SFTSV. AI also constructs B cell lineage trees to trace mAb evolution, pinpointing key mutations that enhance neutralizing activity. Furthermore, AI predicts selection pressures on these mutations, identifying variants with “evolutionary flexibility” for optimization. Analysis of CDR3 lengths and amino acid composition preferences in BCRs informs the design of synthetic mAb libraries and guides the introduction of beneficial mutations to enhance antibody affinity and breadth.
Nanobodies targeting SFTSV offer excellent stability and tissue penetration, but their camelid origin may trigger immune rejection and antidrug antibody (ADA) production. Traditional humanization methods, such as CDR grafting, are challenging due to difficulties in selecting suitable framework regions (FWRs) and maintaining affinity. AI-powered protein structure prediction models like AlphaFold3 have improved humanization by predicting precise 3D structures and analyzing CDR conformations, key FWR residues, and immunogenic epitopes (Bryant et al., 2022). AI can identify optimal human frameworks for CDR grafting and predict the effects on mAb affinity. Tools like DeepImmuno (Li et al., 2014), OptMAVEn (Li et al., 2014), and AbImmPred (Wang et al., 2024) help minimize immunogenicity while preserving functionality. By integrating multiple factors, including humanization degree, immunogenicity, and affinity, AI streamlines the humanization process, reducing experimental costs and accelerating SFTSV nanobody optimization. AI-driven humanization holds promise for resolving the affinity loss associated with traditional approaches.
The high mutation rate of SFTSV poses a challenge to developing broad-spectrum and long-lasting mAbs. AI can analyze the virus genome to identify highly variable regions and potential antigenic drift sites. By employing multiple sequence alignment and temporal sequence analysis, AI can pinpoint mutation hotspots and track their evolutionary trends. Predictive modeling using deep learning models like Bidirectional Long Short-Term Memory networks (Xuan et al., 2019) can identify mutation sequences with high adaptability and immune escape potential. These predictions can be validated using pseudovirus systems and neutralization assays. The findings can inform the design of next-generation broad-spectrum neutralizing mAbs. In addition, AI can help develop proactive defense strategies by optimizing conserved viral epitopes for multivalent vaccines and broad-spectrum mAbs. By integrating genomic surveillance and regional control networks, AI can provide a globally responsive solution for monitoring and intervening in SFTSV outbreaks (Ma et al., 2024).
AI has great potential in SFTSV mAb development, facilitating broad-spectrum antibody screening, synthetic library design, mAb humanization, and viral mutation analysis. While data limitations pose challenges, AI can improve mAb development by optimizing models, enhancing data sharing, and integrating experimental validation.
Discussion
The SFTSV envelope glycoproteins Gn and Gc form higher-order assemblies with icosahedral symmetry (Du et al., 2023), and their N-linked glycans play a crucial role in viral attachment to host cells via C-type lectin receptors like DC-SIGN. The N63 glycosylation site on Gn is conserved across SFTSV genotypes and is targeted by mAb 40C10 and JK-8 through hydrogen bonding (Wu et al., 2024; Zhang et al., 2025). It is hypothesized that these mAbs neutralize SFTSV by blocking Gn-DC-SIGN interactions, but this mechanism requires further validation through competitive binding assays or viral adhesion inhibition studies.
SFTSV glycoproteins can utilize DC-SIGN to enter dendritic cells and potentially disrupt B cell function, impairing the generation of neutralizing mAbs. This may limit the success of single B cell sorting for isolating antigen-specific B cells (Suzuki et al., 2020). To overcome this, convalescent or vaccinated donors can be selected to enhance the likelihood of successful mAb discovery. Fluorescence-labeled SFTSV Gn or Gc proteins can be used with FACS to isolate target cells, and high-throughput sequencing techniques like scRNA-seq or BCR-seq can profile the antibody repertoire. If needed, short-term in vitro activation of B cells with stimulatory factors like IL-21, CD40L, and Cytosine–phosphate–Guanine Oligodeoxynucleotide (CpG ODN) can enhance B cell viability and improve mAb generation (Buchanan et al., 2011; Kuchen et al., 2007; Possamaï et al., 2021).
MAbs can potentially induce ADE of infection if they bind to the virus without neutralizing it, facilitating entry into host cells via Fc receptor-mediated pathways (Wells et al., 2025). ADE is a concern in vaccine and mAb therapy development, as seen in dengue virus infection (Halstead, 1988; Katzelnick et al., 2017; Narayan and Tripathi, 2020). While ADE has been observed in vitro and in animal models for Severe Acute Respiratory Syndrome (SARS)-CoV-2, its clinical relevance is uncertain (Lee WS et al., 2020; Li et al., 2021a). Research on ADE in SFTSV is limited, but given the potential risk, it is crucial to monitor ADE during neutralizing mAb development. Validation studies in transgenic mouse and nonhuman primate models expressing human FcγRs can help assess this risk.
Sequence analysis of SFTSV-neutralizing mAbs revealed distinct structural features associated with high-affinity binding (Table 2 and Fig. 3). MAbs targeting Gn often have extended CDR-H3 regions with aromatic and positively charged residues, enhancing interactions with Gn’s negatively charged surface. Conserved motifs like “GYF” and “IFPG” in CDR-H1 and CDR-H2 are crucial for antigen recognition and should be retained during optimization. The “QSV” motif in CDR-L1 is also characteristic of Gn-specific mAbs. MAbs targeting Gc, such as SF83, typically have compact CDR structures that enable optimal binding to spatially restricted epitopes (Zhang et al., 2025). This structural feature is crucial for high specificity and binding affinity. Therefore, designing Gc-targeting mAbs requires careful consideration of the binding interface’s stereochemical complementarity to the Gc antigen’s constrained spatial configuration.
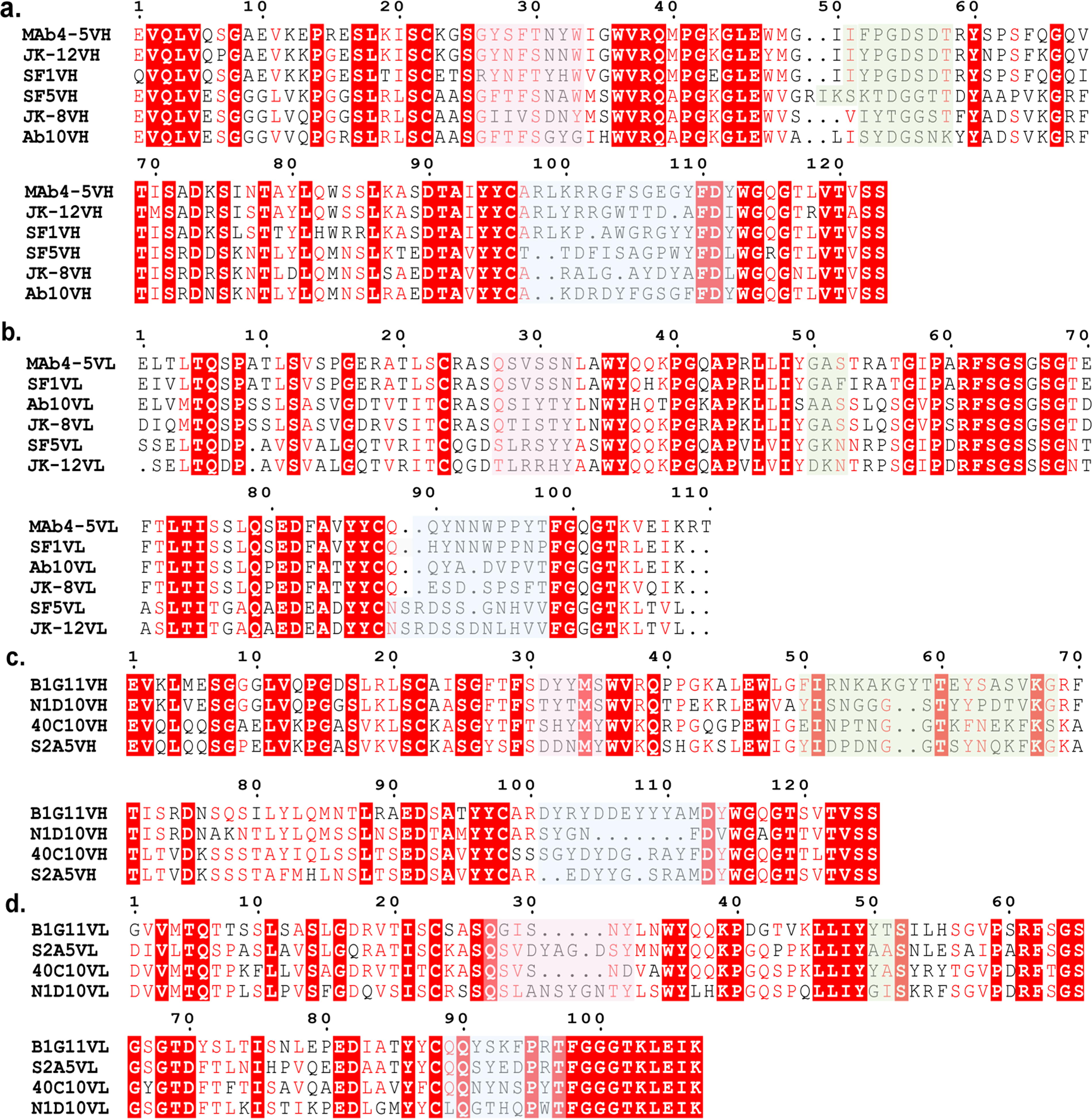
Variable regions of heavy and light chains from published human and murine antibodies targeting SFTSV.
CDR of SFTSV Antibodies
Conclusions
Recent advances in SFTSV mAb development have been driven by technologies such as hybridoma, phage display, single B cell sequencing, and AI-based design. This review highlights the structural and functional characteristics of SFTSV envelope glycoproteins Gn and Gc and summarizes potent neutralizing mAbs. AI-assisted techniques are facilitating mAb engineering, but challenges remain, including limited epitope accessibility and the risk of ADE. To overcome these challenges, future efforts should focus on integrating structure-guided design with high-throughput screening, validating efficacy in nonhuman primate models, and leveraging AI-driven humanization strategies. A multidimensional approach combining structural biology, bioengineering, and AI is essential to develop safe and effective mAb therapies against SFTSV.
Footnotes
Authors’ Contributions
W.Q.L. and M.S.Q. contributed to the conceptualization of the review. W.Q.L. wrote the original draft. M.S.Q. (corresponding author) acquired funding and was responsible for writing—review and editing. Y.L. supervised the project. B.Y.Z. prepared Figure 1. Z.G. prepared ![]() . Z.X. and L.K.J. prepared the table. All authors reviewed and approved the final article. All the authors read and approved the article.
. Z.X. and L.K.J. prepared the table. All authors reviewed and approved the final article. All the authors read and approved the article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
M.S.Q., W.Q.L., Y.L., B.Y.Z., Z.G., and L.K.J. were financially supported by National Natural Science Foundation of China (81772203 [Y.L.]), Key Project of Anhui Institute of Translational Medicine (2021zhyx-B02), and Basic and Clinical Collaboration Enhancement Plan of Anhui Medical University (2022xkjT014).