
Abstract
The cyanobacterial circadian oscillator, consisting of KaiA, KaiB, and KaiC proteins, drives global rhythms of gene expression and compaction of the chromosome and regulates the timing of cell division and natural transformation. While the KaiABC posttranslational oscillator can be reconstituted in vitro, the Kai-based oscillator is subject to several layers of regulation in vivo. Specifically, the oscillator proteins undergo changes in their subcellular localization patterns, where KaiA and KaiC are diffuse throughout the cell during the day and localized as a focus at or near the pole of the cell at night. Here, we report that the CI domain of KaiC, when in a hexameric state, is sufficient to target KaiC to the pole. Moreover, increased ATPase activity of KaiC correlates with enhanced polar localization. We identified proteins associated with KaiC in either a localized or diffuse state. We found that loss of Rbp2, found to be associated with localized KaiC, results in decreased incidence of KaiC localization and long-period circadian phenotypes. Rbp2 is an RNA-binding protein, and it appears that RNA-binding activity of Rbp2 is required to execute clock functions. These findings uncover previously unrecognized roles for Rbp2 in regulating the circadian clock and suggest that the proper localization of KaiC is required for a fully functional clock in vivo.
Circadian rhythms, regulated by a 24 h biological clock, enable the coordination of temporal programs of cellular physiology and facilitate adaptation to daily environmental changes in diverse organisms (Bell-Pedersen et al., 2005). Cyanobacteria are currently the only prokaryotic system in which the molecular details of the circadian clock have been elucidated, with Synechococcus elongatus PCC 7942 serving as the premier model system for the study of the cyanobacterial circadian clock (Kondo et al., 1993). In S. elongatus, a core oscillator encoded by the kaiA, kaiB, and kaiC genes regulates global patterns of gene expression (Ishiura et al., 1998; Kondo et al., 1994), compaction of the chromosome (Smith and Williams, 2006; Woelfle et al., 2007), and the timing of cell division (Cohen and Golden, 2015; Dong et al., 2010; Mori et al., 1996) and natural transformation (Taton et al., 2020). KaiC is a hexameric protein consisting of CI and CII domains that possess autokinase and autophosphatase (CII) and ATPase activities (both) (Nishiwaki et al., 2004; Xu et al., 2004). Rhythmic associations of KaiA and KaiB with KaiC drive ~24 h rhythms of KaiC phosphorylation and dephosphorylation on neighboring serine 431 and threonine 432 residues located in the CII domain (Nishiwaki et al., 2004; Xu et al., 2004). During the day, KaiA associates with the A-loops of KaiC, found on the CII domain, promoting KaiC’s autokinase activity (Kim et al., 2008). Once in a fully phosphorylated state, a KaiB binding site is exposed on the CI domain of KaiC (Chang et al., 2011, 2015). In addition, KaiB must switch from a tetrameric ground-state fold to a monomeric fold-switched form that is competent to bind to KaiC (Chang et al., 2015). Once both these criteria are met, KaiB binds to KaiC and sequesters KaiA in an autoinhibited state, triggering the autophosphatase activity of KaiC at night (Chang et al., 2015; Tseng et al., 2017). Remarkably, these oscillations of KaiC phosphorylation can be reconstituted in vitro solely with purified KaiA, KaiB, KaiC, and ATP (Nakajima et al., 2005).
These endogenously generated rhythms are synchronized with the environment through an input pathway that monitors changes in cellular redox. Specifically, CikA, which also plays key roles in circadian output, and KaiA directly bind oxidized quinones, whose redox status varies as a function of photosynthetic activity, and signal the onset of darkness (Kim et al., 2012; Wood et al., 2010). In addition, a drop in the ATP:ADP ratio in the cell can reset the phase of KaiC phosphorylation directly and functions to signal the duration of the dark period in S. elongatus (Rust et al., 2011). Clock-controlled activities are regulated by histidine kinase SasA and its cognate response regulator RpaA, as well as CikA, which also functions as a phosphatase on RpaA (Gutu and O’Shea, 2013; Takai et al., 2006). Association with KaiC or the KaiABC ternary complex promotes the activities of SasA and CikA, respectively, resulting in rhythmic phosphorylation of RpaA (Gutu and O’Shea, 2013), which in turn promotes global rhythms of gene expression (Markson et al., 2013) and the timing of cell division (Mori et al., 1996; Yang et al., 2010).
While several aspects of the clock can be reconstituted in vitro, including rhythms of KaiC phosphorylation (Nakajima et al., 2005) as well as RpaA phosphorylation and rhythmic DNA binding (Chavan et al., 2021), the clock in vivo is subject to many additional layers of regulation. Rhythms of KaiB and KaiC protein abundance are observed, where peak expression is achieved after dusk (Kitayama et al., 2003), and the overexpression or underexpression of any of the kai genes results in loss of rhythmicity (Ishiura et al., 1998; Xu et al., 2013). Moreover, the clock undergoes an elegant reorganization in its subcellular localization where KaiA and KaiC are diffuse throughout the cell during the day and become highly localized as discrete foci near a single pole of cells at night, in a clock-dependent fashion (Cohen et al., 2014). KaiA localization is dependent on KaiC, and KaiA and KaiC colocalize with CikA at night (Cohen et al., 2014). CikA is constitutively localized to the cell pole, where polar localization is observed at all circadian times (Cohen et al., 2014; Zhang et al., 2006). Although it is not understood how or why the clock proteins exhibit these rhythms in their subcellular localization patterns, these movements have been proposed to contribute to the robustness and synchronization of the circadian clock.
Here, we report that the CI domain of KaiC is sufficient to support KaiC localization to cell poles and that the ATPase activity of KaiC correlates with the extent of polar localization. We identified proteins that associate specifically with either cytoplasmic or localized mutant variants of KaiC. Specifically, we focus on the RNA-binding protein, Rbp2. We found that deletion of rbp2 results in a long circadian period and reduced polar localization of KaiC, demonstrating that Rbp2 represents a previously unidentified component of the extended clock network. In addition, we demonstrate that Rbp2 is the only RNA recognition motif (RRM) domain-containing protein encoded in the S. elongatus genome to be involved in clock regulation. We present evidence that the RNA-binding activities of Rbp2 are critical for the role that Rbp2 plays in regulating the clock. The results suggest that new components of the extended clock network that are important for clock function in vivo can be identified and reveal previously unrecognized roles for an RNA-binding protein in regulating clock function.
Materials and Methods
Bacterial Strains, Growth Conditions, and DNA Manipulations
Plasmids and Escherichia coli and S. elongatus strains are described in Supplementary Tables S1 and S2. S. elongatus strains were grown as previously described (Clerico et al., 2007). Briefly, strains were grown in BG-11 media with the appropriate antibiotics (Taton et al., 2014). Antibiotics used in this study include chloramphenicol, kanamycin, spectinomycin and streptomycin (Sp and Sm), gentamycin, and nourseothricin; concentrations were used as previously described (Taton et al., 2014). Plasmids were designed using the CYANO-VECTOR assembly portal (http://golden.ucsd.edu/CyanoVECTOR/), constructed with the GeneArt Seamless Cloning and Assembly Kit (Life Technologies), and propagated in E. coli XL1 Blue cells as previously described (Taton et al., 2014). Complementation and expression strains were constructed by expressing genes in one of three S. elongatus neutral sites (NS) NS1, NS2, or NS3. For the construction of knockout strains, complete segregation of the mutant loci was verified by PCR. Point mutants were constructed using the QuickChange (Stratagene California) protocol, with clones verified by Sanger sequencing.
Immunoprecipitation and Mass Spectrometry
S. elongatus cells (5 × 109) were harvested and lysed via bead beating as previously described (Ivleva and Golden, 2007) in buffer containing phosphate-buffered saline (PBS), 1 mM phenylmethylsulphonyl fluoride (PMSF), and protease inhibitor cocktail (Roche). Rabbit IgG polyclonal Green Flourescent Protein (GFP) antibody (Life Technologies) was conjugated to magnetic Protein G Dynabeads (Thermo) following the instructions of the manufacturer and incubated with the extract for 4 h at 4 °C with rotation. The beads were washed 4 times in Interactants (IA) buffer, 50 mM NaH2PO4 pH 7.8, 5 mM NaCl, 0.1% TritonX-100, 1 mM PMSF, 15 U/mL DNaseI, protease inhibitor cocktail (Roche), and PhosSTOP phosphatase inhibitor (Roche), and 3 times with PBS. Protein complexes were eluted in 50 mM glycine pH 2.8.
Protein samples were diluted in Tris ETDA NaCL (TNE) (50 mM Tris pH 8.0, 100 mM NaCl, 1 mM EDTA) buffer. RapiGest SF reagent (Waters Corporation) was added to the mix to a final concentration of 0.1%, and samples were boiled for 5 min. Tris(2-carboxyethyl) phosphine (TCEP) was added to 1 mM (final concentration), and the samples were incubated at 37° C for 30 min. Subsequently, the samples were carboxymethylated with 0.5 mg/ml of iodoacetamide for 30 min at 37 °C followed by neutralization with 2 mM TCEP (final concentration). Protein samples prepared by the aforementioned steps were digested with trypsin (trypsin:protein ratio = 1:50) overnight at 37 °C. RapiGest was degraded and removed by treating the samples with 250 mM HCl at 37 °C for 1 h, followed by centrifugation at 12,000 × g for 30 min at 4 °C. The soluble fraction was then added to a new tube, and the peptides were extracted and desalted using C18 desalting columns (Thermo Fisher Scientific, PI-87782). Peptides were quantified using Bicinchoninic acid (BCA) assay, and a total of 1 mg of peptides was injected for liquid chromatography-mass spectrometry (LC-MS) analysis.
Liquid chromatography with tandem mass spectroscopy (LC-MS/MS) analysis was performed as previously described (Guttman et al., 2009; McCormack et al., 1997). Trypsin-digested peptides were analyzed by ultra-high-pressure liquid chromatography (UPLC) coupled with tandem mass spectroscopy (UPLC-MS/MS) using nanospray ionization. The nanospray ionization experiments were performed using a Orbitrap Fusion Lumos Tribrid Mass Spectrometer (Thermo Fisher Scientific) interfaced with nano-scale reversed-phase UPLC (Thermo Dionex UltiMate 3000 RSLCnano System) using a 25 cm, 75-micron ID glass capillary packed with 1.7 µm C18 (130) BEH beads (Waters corporation). Peptides were eluted from the C18 column into the mass spectrometer using a linear gradient (5%-80%) of acetonitrile. Mass spectrometer parameters included an MS1 survey scan using the Orbitrap detector (mass range [m/z]: 400-1500 [using quadrupole isolation], 120,000 resolution setting, spray voltage of 2200 V, ion transfer tube temperature of 275 °C, AGC target of 400,000, and maximum injection time of 50 ms) was followed by data-dependent scans (top speed for most intense ions, with charge state set to only include + 2-5 ions), and 5 sec exclusion time, while selecting ions with minimal intensities of 50,000, in which the collision event was carried out in the high-energy collision cell (HCD collision energy of 30%), and the fragment masses were analyzed in the ion trap mass analyzer (with ion trap scan rate of turbo, first mass m/z was 100, AGC Target 5000, and maximum injection time of 35 ms). Protein identification was carried out using Peaks Studio 8.5 (Bioinformatics solutions Inc.).
Circadian Bioluminescence Monitoring
Bioluminescence was monitored using a P kaiB -luc firefly luciferase fusion reporter inserted into an NS of the S. elongatus chromosome at 30 °C under constant light after 2 entrainment cycles of 12 h in the light followed by 12 h in the dark (LD12:12) to synchronize the population as previously described (Mackey et al., 2007). Bioluminescence was monitored every 2 h using a PerkinElmer TopCount bioluminescence plate reader. Data were collected and plotted using Microsoft Excel. Data from 12 wells of each genotype were analyzed for rhythmicity, including period ± standard deviation, using the Biological Rhythms Analysis Software System (BRASS) within Microsoft Excel.
Fluorescence Microscopy and Image Analysis
Cells were placed on a pad of 1.2% agarose in BG-11 medium and covered with a coverslip. Microscopy was performed with a DeltaVision Core system (Applied Precision) with a WeatherStation attached to an Olympus IX71 inverted microscope and an Olympus Plan Apochromat 100× objective at 30 °C with tetramethyl rhodamine isocyanate (EX555/EM617) and Yellow Fluorescent Protein (YFP) (EX500/EM535) filter settings. Images were captured using a CoolSnap HQ CCD camera (Photometrics) and deconvolved using the SoftWorx imaging program (Applied Precision). Exposure times were limited to conditions under which we do not observe fluorescence from wild type (WT) strains in the YFP channels to limit bleed-through from thylakoid fluorescence (Cohen et al., 2015). For analysis of KaiC localization in time-course experiments, aliquots of cells were taken at designated time points and fixed directly in BG-11 growth medium with a final concentration of 2.4% (vol/vol) paraformaldehyde (Electron Microscopy Sciences) in 30 mM NaPO4 buffer (pH 7.5) for 20 min at room temperature before they were moved to 4 °C. Images were colorized in SoftWorx and then transferred to Photoshop (Adobe) for figure assembly. KaiC foci tracking was performed as previously described (Cohen et al., 2014). For imaging E. coli, aliquots of cells were stained with the membrane dye FM4-64 (240 ng/ml-1 µg/ml; Molecular Probes) and 4,’6-diamidino-2-phenylindole (0.2 µg/mL; Thermo Fisher Scientific) prior to imaging.
Generation of Homology Model
An Rbp2 homology model was generated with SWISS-Model based on the human postcatalytic splicesome (P complex) PDB 61CZ (Zhang et al., 2019). Initial template selection from automatically generated models were based on 3 criteria: minimum of 30% sequence identity (Haddad et al., 2020), conservation of key Ribonucleo protein (RNP) residues, and the known structure of 4 beta-sheets and 2 alpha-helices for RRM domain proteins (Maris et al., 2005). The selected homology model was then input into GalaxyRefine2 for energy minimization of the protein fold via the Critical Assessment of Structure Prediction (CASP) algorithm (Lee et al., 2018, 2019). The refined homology model was selected from the 10 likeliest proposed models based on scores including energy, MolProbity validation, root mean squared deviation, clash score, number of poor rotamers, and Ramachandran score (Lee et al., 2018, 2019). RNA was manually modeled via PyMOL. The RNA sequence was determined based on the likeliest interaction of RNA bases and corresponding amino acids (Kligun and Mandel-Gutfreund, 2015).
Protein Purification and Gel Filtration Analysis
E. coli XL1 blue strain carrying pLA0004 was used to overexpress Strep-tagged Rbp2 (Rbp2-Strep) protein from a P trc promoter. A 20-ml overnight E. coli starter culture in LB medium was transferred to 1 L of Luria-Bertani (LB) medium supplemented with 20 μg/ml Sp plus 20 μg/ml Sm and grown to OD600 ~0.4 at 37 °C before induction with 0.2-mM isopropyl β-D-1-thiogalactopyranoside (IPTG) overnight at 22 °C. E. coli cells were collected by centrifugation and re-suspended in Strep-Tactin wash buffer (50 mM Tris pH 8.0, 150 mM NaCl, 5% glycerol) before cell lysis with an Avestin C3 Emulsiflex homogenizer (Avestin Inc, Canada). Clarified cell lysate was loaded onto a Strep-Tactin XT Superflow (IBA Lifesciences) gravity column equilibrated with Strep-Tactin wash buffer. The column was then washed with Strep-Tactin wash buffer, followed by protein elution with the same buffer containing 50 mM biotin. Rbp2-containing eluents were concentrated using spin concentrators and loaded onto a prep-grade Superdex 200 column equilibrated in a buffer containing 50 mM Tris pH 8.0, 150 mM NaCl, and 5% glycerol for further purification with gel filtration chromatography. The purification of KaiC was performed as previously reported (Tseng et al., 2017).
Binding reactions were set up with different combinations of 25 μM KaiC, Rbp2, or poly(rU) (20-mer ordered from Integrated DNA Technologies) in clock buffer (20 mM Tris pH 8.0, 150 mM NaCl, 5 mM MgCl2, 0.5 mM EDTA, and 1 mM ATP) at 30 °C. After 30 min incubation, 100 μl of the reaction mix was injected to an analytical-grade Superdex 200 column equilibrated with the same clock buffer for gel filtration chromatography at room temperature with 1 ml/min flow rate.
Results
CI Domain of KaiC Is Sufficient to Support KaiC Polar Localization
In order to investigate whether a particular domain of KaiC is necessary to support polar localization, we expressed YFP-fusions to either the full-length as well as isolated CI or CII domains of KaiC. Previous studies demonstrated that a YFP-KaiC fusion protein is functional and expressed as a full-length protein (Cohen et al., 2014; Figure 1a). We observed that expression of the CI domain alone supports polar localization, whereas expression of the CII domain alone does not, as diffuse localization was observed (Figure 1b and 1c). When the CII domain only was overexpressed from the P trc promoter by the addition of inducer IPTG, 6% of cells showed polar localization compared to 96% of cells for the full-length KaiC construct. As the CI and CII domains of KaiC are known to form hexamers in vitro (Chang et al., 2011), we aimed to determine if the CI domain of KaiC is in a hexameric state when localized to the cell poles. We introduced mutations that substitute Ala for Arg at position 40 (R40A) and for Lys at position 172 (K172A), known to result in monomeric CI domain proteins in vitro (Tseng et al., 2013), into the YFP-CI domain fusion protein. We observed that monomeric CI domain of KaiC was unable to support polar KaiC localization (Figure 1d), supporting the notion that the CI domain of KaiC needs to be in a hexameric form to be able to localize to the cell pole.
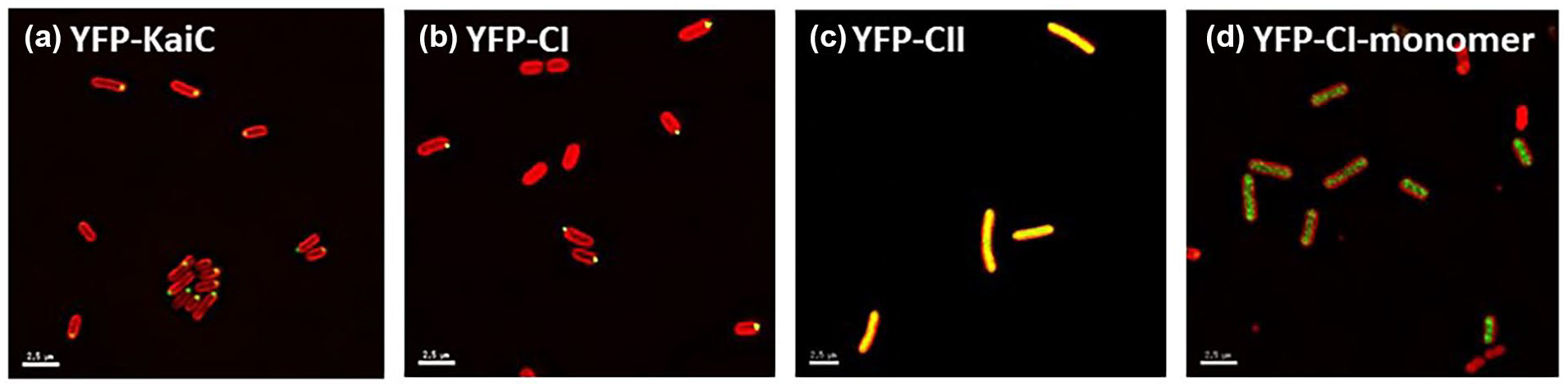
The CI domain of KaiC is sufficient to support KaiC polar localization. Fluorescent micrographs of cells expressing (a) YFP-KaiC full-length fusion, (b) YFP-KaiC-CI domain fusion, (c) YFP-KaiC-CII domain fusion, and (d) YFP-KaiC-CIR40A-K172A monomer fusion (yellow) in a ΔkaiC background (AMC704) shows that the CI domain alone, when in a hexameric state, can localize to the cell pole while the CII domain is not capable of polar localization. When the YFP-KaiC-CII domain only construct was overexpressed, only 6% of the cells showed polar localization compared to 96% of cells in the full-length YFP-KaiC fusion. Autofluorescence is shown in red. Scale bars = 2.5 microns. Abbreviation: YFP = Yellow Fluorescent Protein.
High ATPase Activity Correlates with Enhanced KaiC Polar Localization
The CI domain of KaiC possesses ATPase activity that is separate from the ATPase in the CII domain required for autophosphorylation, and both domains are required for a functional clock. Although the ATPase is weak, consuming only about 15 molecules of ATP per day, it is temperature-compensated, and its activity oscillates in the presence of KaiA and KaiB (Terauchi et al., 2007). Approximately 70% of KaiC ATPase is attributable to the CI domain (Murakami et al., 2008; Terauchi et al., 2007). It is proposed that the CI ATPase activity is important for inducing structural changes that regulates the timing and recruitment of KaiB (Mutoh et al., 2013). Moreover, this ATPase activity of KaiC is correlated with the circadian period, leading to the hypothesis that reduced ATPase activity results in longer circadian periods and enhanced ATPase activity results in shorter circadian periods (Terauchi et al., 2007). As the CI domain of KaiC is sufficient to promote KaiC polar localization, we sought to determine whether the ATPase activity of KaiC correlates with polar localization. We tested this hypothesis by making several mutations known to alter the ATPase activity of KaiC (Dong et al., 2010; Terauchi et al., 2007) within the full-length KaiC fluorescent fusion and assessing KaiC localization at Zeitgeber time (ZT) 20, representing 8 h after the onset of darkness. These reporter strains were sampled in a light:dark cycle, rather than in constant light, because many of the mutant variants have altered or nonfunctional circadian clocks. Specifically we tested 7 mutant strains whose ATPase activity is altered compared to the WT. KaiC localization was compared among mutant variants A251 V and T42 S, which have longer circadian periods and reduced ATPase activity than WT, and S157 P, F470Y, and R393 C, which have shorter circadian periods and elevated ATPase activity than WT, (Terauchi et al., 2007); KaiC-AA, in which mutation of phosphorylation sites S431 and T432 to alanine results in arrhythmia (Nishiwaki et al., 2004) and elevated ATPase activity (Terauchi et al., 2007); CI, catalytic mutant E77Q and E78Q which exhibits arrhythmia (Phong et al., 2013) and decreased ATPase activity (Kitayama et al., 2013); and WT. We observed that higher ATPase activity is correlated with higher incidence of KaiC polar localization (Figure 2). The KaiC ATPase mutants used represent changes to the combined activity of the CI and CII domains. Thus, we can conclude that KaiC polar localization is correlated with ATPase activity of KaiC, but not necessarily with CI-specific ATPase activity.
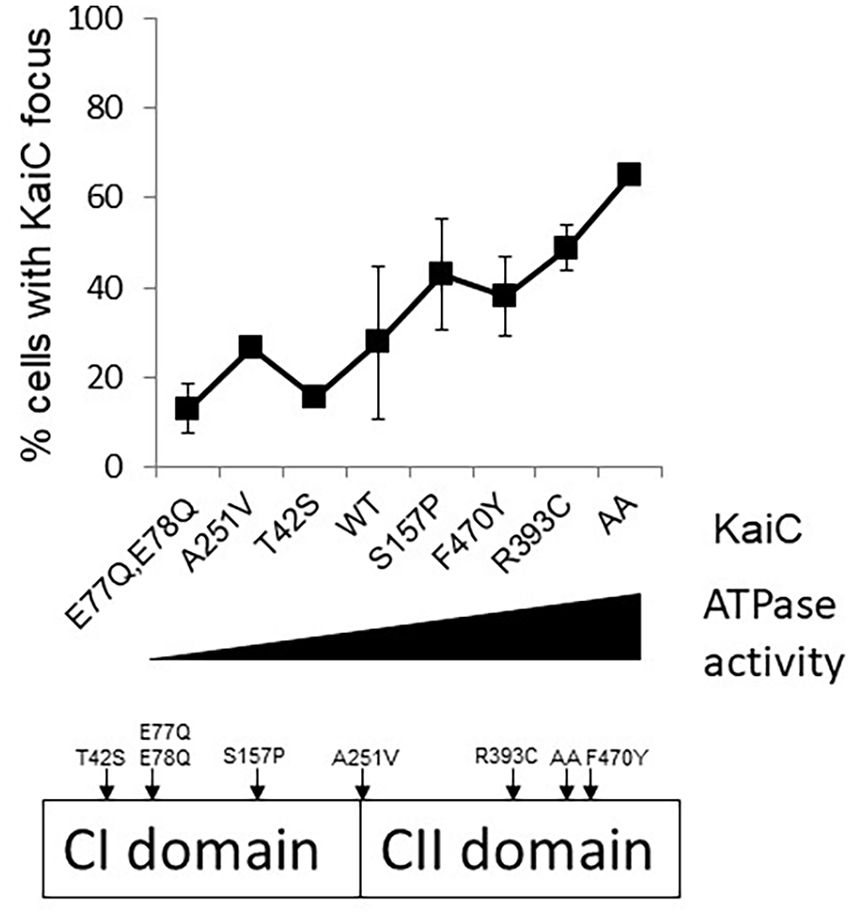
Increased ATPase activity is correlated with enhanced KaiC polar localization. Percentage of cells containing a KaiC focus (y-axis) increases as ATPase activity increases (x-axis). Varying levels of ATPase activity, taken from the literature, are plotted from lowest (left) to highest (right) on the x-axis. KaiC mutant variants were expressed in a ΔkaiC background (AMC704). Strains were entrained in 12 h light:12 h dark, and samples were collected at ZT20, 8 h after the onset of darkness. Percentage of cells with foci was calculated from at least 100 cells from 3 frames. Numbers represent average ± the standard deviation from 3 independent experiments. This schematic shows the positions of ATPase mutations used on the KaiC protein.
Identification of Proteins That Interact with KaiC in a Specific Localization State
Although a wide variety of cellular components in bacteria are targeted to cell poles, including those involved in chromosome segregation, chemotactic signaling, and motility, the mechanisms that establish polar recognition remain poorly understood (Davis and Waldor, 2013; Laloux and Jacobs-Wagner, 2013). Similarly, the mechanism that drives rhythms of KaiC polar localization remains elusive although we previously determined that polar localization is not due to the formation of inclusion bodies or recruitment by the ClpXP protease (Cohen et al., 2014). Moreover, we determined that polar localization is not determined by nucleoid occlusion, membrane potential, or recruitment by polar coordinators/anchors/transporters or the by the Min system (Figure 3). We previously showed that the mechanism of KaiC localization is conserved in E. coli, as S. elongatus KaiC localizes to the pole when expressed in E. coli (Cohen et al., 2014). To determine whether nucleoid occlusion is involved in KaiC localization, we used a strain of E. coli expressing the dnaN159 temperature-sensitive allele. When shifted to the restrictive temperature, the chromosome condenses, creating large nucleoid-free regions (Sutton, 2004). We found that KaiC remains localized as discrete foci even when large nucleoid-free regions are induced (Figure 3a and 3b), indicating that nucleoid occlusion is not playing a role in promoting KaiC localization. We also tested whether membrane potential is involved in the subcellular localization of KaiC by treating S. elongatus cells with either pore former Nisin (Wiedemann et al., 2001) or the uncoupling agent 2,4-dinitrophenol (Gage and Neidhardt, 1993); in both cases, KaiC remained localized to the cell poles (Figure 3c-3e). We also tested KaiC polar localization in mutants deleted for SynPCC7942_1816 or SynPCC7942_1310, which are homologues of E. coli genes that encode proteins that we named polar CATs (Coordinators/Anchors/Transporters) because they positively affect localization of other proteins to the cell poles (see Suppl. Text and Suppl. Figs. S1 and S2). KaiC remained at the poles in cells deleted for these genes (Figure 3f and 3g), implying that they are not important for its localization to the pole. Finally, we sought to determine whether the Min system is involved in KaiC polar localization by assaying for KaiC localization in various min mutants. We observed that KaiC remains localized to the cell poles when minC (Figure 3h), minD1 (Figure 3i), minD2 (Figure 3j), or both minD1 and minD2 are deleted (Figure 3k), indicating that the Min system is not involved in localizing KaiC to the poles of cells.
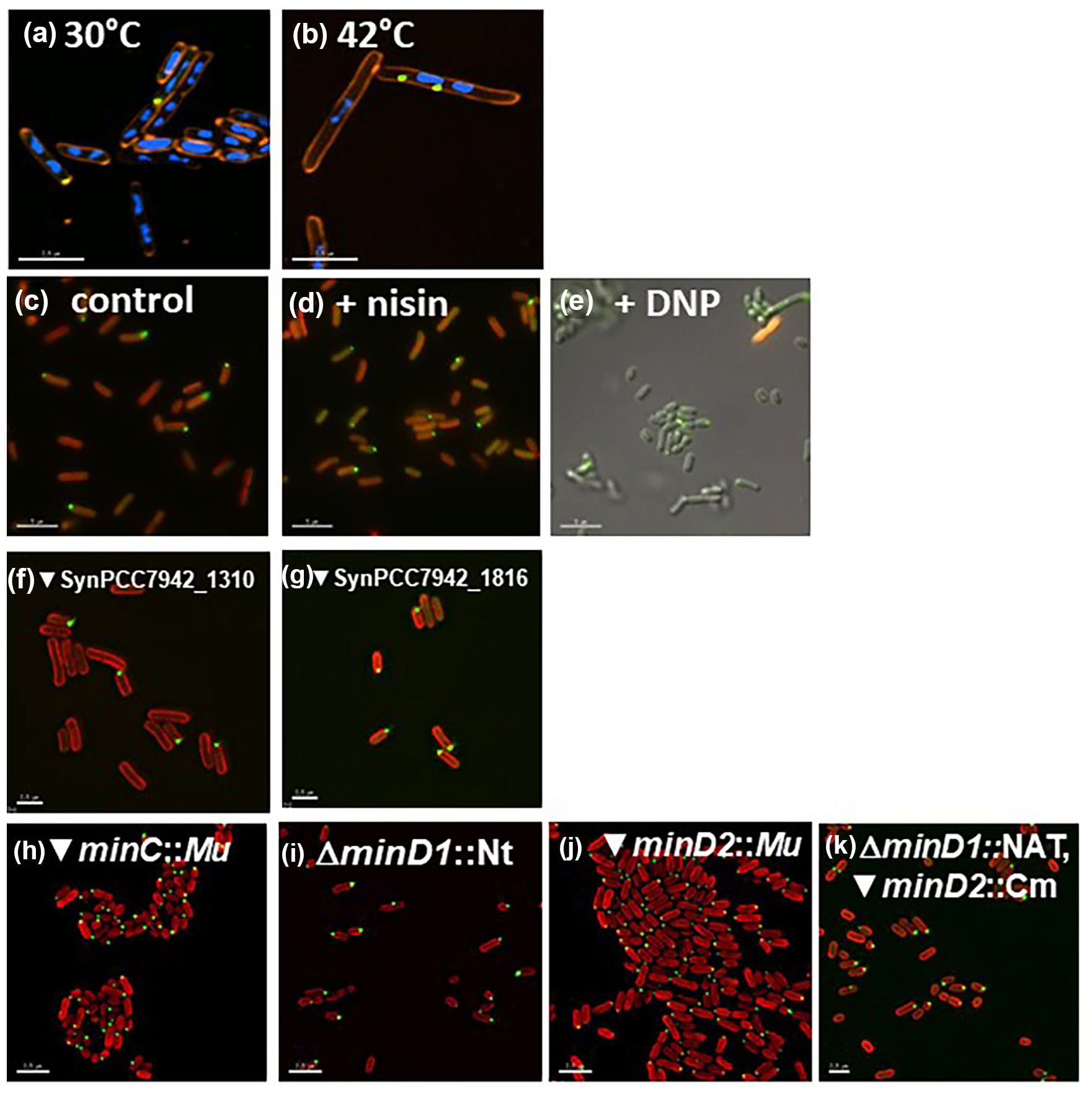
KaiC polar localization is not determined by various cellular factors. (a, b) KaiC localization is not driven by nucleoid occlusion. YFP-KaiC (green) was expressed in the temperature-sensitive E. coli strain AB1157 dnaN159, which was diluted into fresh LB medium and (a) kept at permissive temperature (30 °C) or (b) shifted to the nonpermissive temperature (42 °C) for 2 h to create a large nucleoid-free region. Cells were stained with vital membrane FM4-64 (red) and 4,’6-diamidino-2-phenylindole (blue) to stain the DNA. Even when a large nucleoid-free region was created, KaiC still localized as a discrete focus, indicating that nucleoid occlusion does not drive KaiC localization. Scale bar = 2.5 microns. (c, d) KaiC localization is not driven by membrane potential. S. elongatus cells treated with either (c) 0.025% DMSO control, (d) 1.25 µg/mL pore former Nisin, or (e) 184 µg/mL chemical uncoupler 2,4-dinitrophenol (DNP) show that KaiC remains localized even after membrane potential is perturbed. Autofluorescence is shown in red, except in panel (e) because treatment with DNP resulted in loss of autofluorescence; therefore, KaiC localization in (e) is overlaid onto the differential interference contrast (DIC) image. Scale bar = 5 microns. (f, g) Polar CATs are not responsible for KaiC localization. KaiC remains localized in strains in which homologs of E. coli polar CATs have been mutated, (f) SynPCC7942_1310 and (g) SynPCC7942_1816. Autofluorescence is shown in red. Scale bar = 2.5 microns. (h-k) The Min system is not responsible for KaiC localization. KaiC remains localized in (h) minC insertional mutant, (i) minD1 deletion mutant, (j) minD2 insertional mutant, and (k) minD1, minD2 double mutant. Autofluorescence is shown in red. Scale bar = 2.5 microns. Abbreviations: CATs = Coordinators/Anchors/Transporters; YFP = Yellow Fluorescent Protein.
In order to discover unrecognized mechanisms of KaiC localization, we took an unbiased approach by searching for interacting proteins through immunoprecipitation and mass spectrometry. We sought to identify factors that might be required to either target KaiC to the pole at night or sequester KaiC in the cytosol during the day, by identifying proteins that interact with KaiC, preferentially in either a localized or a diffuse state through the use of mutant variants. KaiC-AA, which carries a substitution of the rhythmically phosphorylated serine and threonine with alanine, has high ATPase activity and displays constitutive polar localization, whereas KaiC-AE, which carries a substitution of the phosphorylated serine and threonine with alanine and glutamic acid, respectively, a phosphomimetic of the phospho-threonine state, is constitutively hypolocalized and found primarily in the cytosol (Cohen et al., 2014). We performed immunoprecipitation experiments by capturing the YFP of the YFP-KaiC fusions, as well as a free YFP control. Mass spectrometry analysis identified 22 proteins that interact specifically with localized KaiC-AA and not with KaiC-AE or free YFP and 7 proteins that interact with diffuse KaiC-AE and not KaiC-AA or free YFP (Suppl. Table S3), for a total of 29 proteins.
From this initial set of 29 proteins, we successfully disrupted genes encoding 11 of them and tested the resulting mutant strains for alterations in rhythms of gene expression or KaiC localization. The remaining targets represent essential genes, and a knockout or insertion mutant could not be generated. We observed that the disruption of 2 genes, SynPCC7942_0417 and SynPCC7942_1999, whose products both associated with localized KaiC-AA, altered both rhythms of gene expression and KaiC localization compared with the WT. SynPCC7942_0417 is an unannotated gene whose sequence suggests that it encodes a member of the AAA + family of ATPases. Disruption of SynPCC7942_0417 led to an approximately 1 hour longer period than the WT (Suppl. Fig. S3) and a mild reduction in KaiC localization at night (Figure 4a). Interestingly, protein localization prediction tools suggest that SynPCC7942_0417 displays cytoplasmic membrane localization, similar to the cell division protease FtsH, which is also a member of the AAA + family of ATPases (Gardy et al., 2003). However, here, we focus on SynPCC7942_1999, which encodes the Rbp2 protein. As neither of these genes has been previously implicated in circadian rhythms, they represent novel avenues for understanding how the clock functions in vivo.
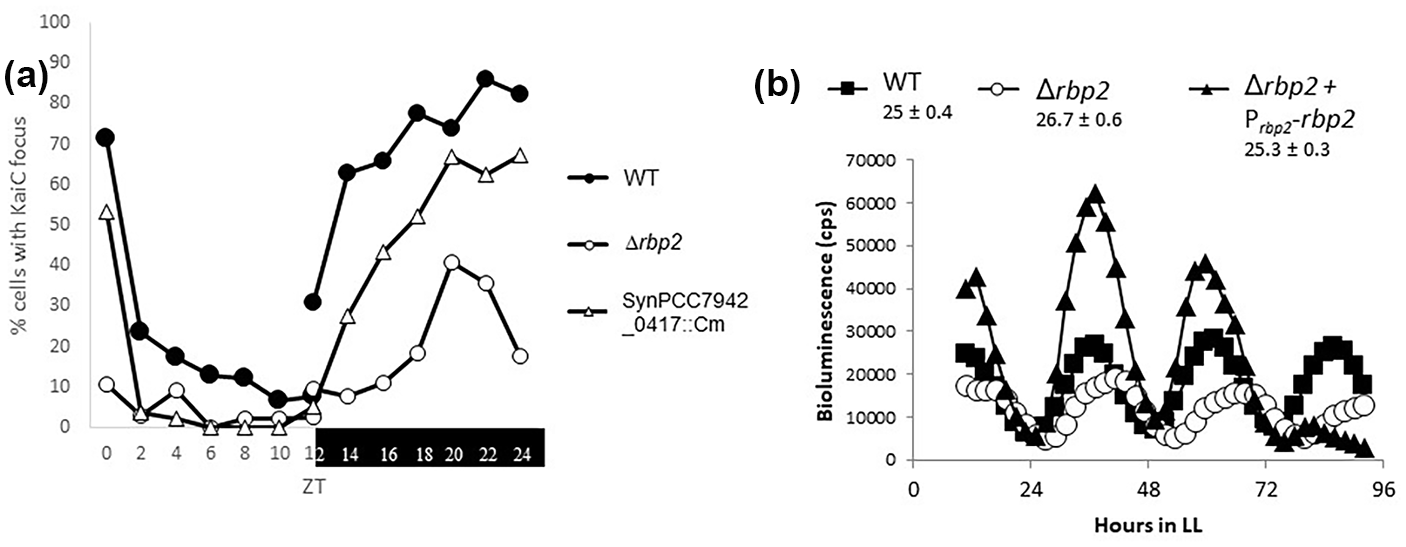
Disruption of rbp2 results in altered circadian phenotypes. (a) Strains expressing YFP-KaiC were entrained to opposite light-dark cycles and sampled every 2 h, with the light (ZT 0-12) and dark (ZT 12-24) samples removed from different incubators at the same laboratory time. ZT refers to the time relative to when the lights turn on. Deletion of rbp2 results in a ~50% reduction (open circles), and disruption of SynPCC7942_0417, a slight reduction (triangles) compared to the WT (closed circles). WT is AMC2158-transformed with pAM5428 (ΔkaiC) and pAM5081 (YFP-KaiC). (b) Bioluminescence output represented as counts per sec from strains carrying PkaiB-luc reporter. LL on x-axis refers to constant light. Deletion of rbp2 results in long-period rhythms of gene expression, open circles (26.7 ± 0.6 h), compared with WT AMC2036, squares (25 ± 0.4 h). This defect can be resolved by expressing rbp2 under its native promoter from NS1, triangles (25.3 ± 0.3 h). Abbreviations: WT = wild type; ZT = Zeitgeber time; YFP = Yellow Fluorescent Protein.
Deletion of rbp2 Results in Changes to the Circadian Clock
Rbp2 is an RNA-binding protein that contains a single RRM. RRM-domain-containing proteins are most commonly found in eukaryotes, with only 85 proteins identified in bacteria, most of which are cyanobacteria (Maruyama et al., 1999). In eukaryotes, RRM-domain proteins are involved in all aspects of posttranslational regulation; however, their roles in bacteria remain unclear (Maris et al., 2005). We found that deletion of rbp2 (Δrbp2) results in an approximate 50% reduction in KaiC localization at night (Figure 4a) and a ~1.5 hour longer period, which can be complemented by expressing rbp2 from an NS under its native promoter (Figure 4b). Taken together, these data suggest that Rbp2 plays a role in regulating the circadian clock and supports the notion that the proper localization of KaiC is important for WT clock function.
Rbp2 Is the Only RRM-Domain Protein Involved in the Circadian Clock
The S. elongatus genome encodes 3 RRM-domain proteins: Rbp1, Rbp2, and Rbp3. Rbp1 consists of an RRM domain followed by a glycine-rich domain, while Rbp2 consists solely of a single RRM domain, and Rbp3 contains an RRM domain followed by a conserved PDPRWA motif in the C-terminus (Figure 5a). The Rbp proteins were initially proposed to have a similar function to the cold-shock RNA chaperones of other bacteria, promoting translation at low temperatures (C. Sugita et al., 1999), because clear homologs of the canonical bacterial cold-shock RNA chaperones are not present in S. elongatus. Indeed, Rbp1 is induced by cold shock and promotes survival under low temperatures (Mutsuda et al., 1999; C. Sugita et al., 1999). However, neither Rbp2 nor Rbp3 is induced by low temperatures, and they do not contribute to cellular survival at low temperatures (Hayashi et al., 2017; C. Sugita et al., 1999). In addition, Rbp1 and Rbp2 have been verified experimentally to bind RNA in vitro (M. Sugita and Sugiura, 1994). While the roles for Rbp1 in promoting survival in response to cold stress seem clear, the functions of Rbp2 (and Rbp3) in the cell remain elusive.
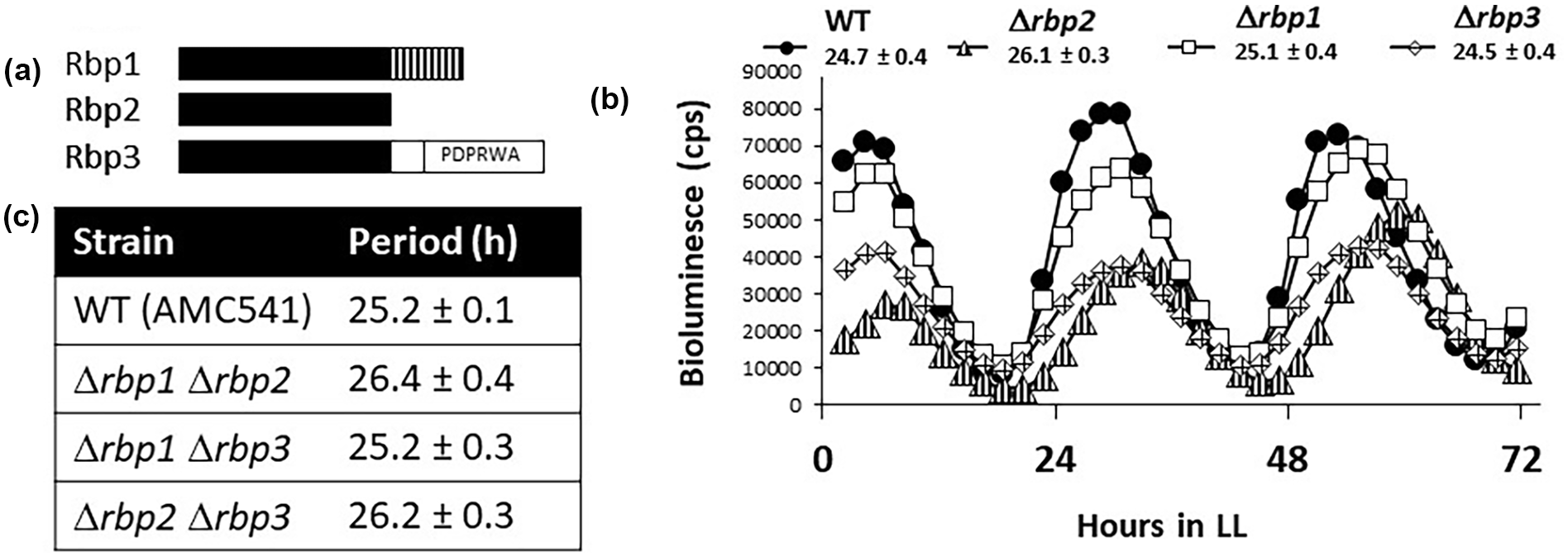
rbp2 Is the only RRM-domain-containing protein that functions in the circadian clock. (a) Schematic representation of the domain structure of the 3 Rbp proteins in S. elongatus. Black bars represent the RRM domain, vertical lines represent the glycine-rich domain found only on Rbp1, and an open box represents the conserved “PDPRWA” motif found at the C-terminus of Rbp3. (b) Bioluminescence output, in counts per sec, from a P kaiB -luc reporter. Deletion of rbp2 results in long-period rhythms of gene expression (triangles, 26.1 ± 0.3 h), whereas rhythms after deletion of rbp1 (squares, 25.1 ± 0.4 h) or rbp3 (diamonds, 24.5 ± 0.4 h) are similar to the WT (closed circles, 24.7 ± 0.4 h). WT is AMC2036. (c) Table reporting the period ± standard deviation of rbp double mutants, calculated from bioluminescence traces from a P kaiB -luc reporter. WT is AMC541. Abbreviations: RNA = ribonucleic acid; RRM = RNA recognition motif; WT = wild type.
In order to determine whether Rbp2 is the only RRM-domain protein involved in clock function, we generated strains that lack each of the rbp genes as well as all combinations of pairwise deletions. We were unable to generate the triple-mutant strain, suggesting that the rbp genes likely have some overlapping function that is required for viability. In addition, we found that while Δrbp1 strains are viable, they could not be maintained for long periods of time. We found that only strains lacking rbp2, both single- and double-mutant strains (Δrbp1Δrbp2 and Δrbp2Δrbp3), displayed any circadian phenotypes, where the loss of rbp2 resulted in a 1-1.5 h longer circadian period of gene expression (Figure 5b and 5c). These data suggest that while the three Rbp proteins may have some overlapping function, Rbp2 is the only RRM-domain protein that is involved in clock function.
RNA-Binding Activity of Rbp2 Is Likely Involved in Regulation of the Circadian Clock
As Rbp2 consists solely of an RRM domain that has been biochemically verified to bind RNA (M. Sugita and Sugiura, 1994), we sought to determine whether the RNA-binding activity is required for its roles in regulating the circadian clock. The RRM domain forms an αβ sandwich structure that consists of subdomains RNP 1, located in the C-terminus, and RNP 2, located in the N-terminus. Four conserved hydrophobic residues in these motifs contribute to RNA binding (Maris et al., 2005). We generated a homology model of Rbp2 based on published structures of eukaryotic RRM domains and found that tyrosine 4, located in RNP 2, and arginine 42, phenylalanine 44, and phenylalanine 46, located in RNP 1, of Rbp2 are similarly poised to coordinate binding to RNA (Figure 6a). We mutated these conserved amino acids to alanine in order to generate strains expressing rbp2-Y4A or rbp2-R42A-F44A-F46A. We found that expression of these mutant variants, predicted to have reduced ability to bind to RNA, does not complement the rbp2 null strain, where long-period circadian rhythms of gene expression are observed (Figure 6b). These data suggest that the RNA-binding activities of Rbp2 are necessary for it to execute its effect on the circadian clock.
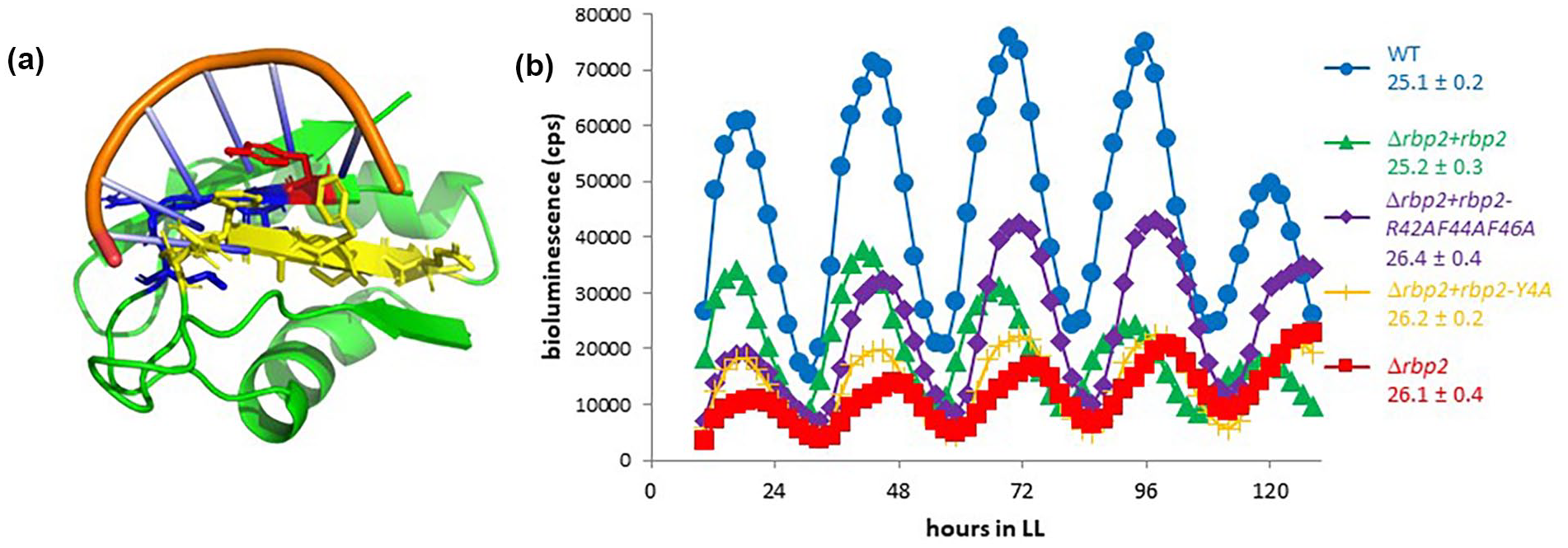
RNA-binding activity of Rbp2 is likely required to execute circadian functions. (a) Homology model of Rbp2 bound to RNA. Rbp2 (green), RNA-binding domains RNP-2 (blue) and RNP-1 (yellow). RNP-1 aromatic residues R42-F44-F46 known to be important for RNA binding and Y4 located in RNP-2. Position Y4 is shown in red. (b) Bioluminescence output, in counts per sec, from a P kaiB -luc reporter. Mutations that likely impair RNA binding rbp2-Y4A (yellow cross) and rbp2-R42A-F44A-F46A (purple diamonds) confer a long-period rhythm of gene expression, similar to a rbp2 deletion (red squares), compared to WT (blue circles) or the complemented strain (green triangles). WT is AMC2036. Abbreviations: RNA = ribonucleic acid; WT = wild type.
Rbp2 and KaiC Likely Interact Indirectly
To determine whether Rbp2 is associating with KaiC directly or indirectly, we performed gel filtration analysis. After a 30 min incubation of purified Rbp2 and KaiC, no Rbp2-KaiC complex was observed, suggesting that these two proteins do not interact directly in vitro (Figure 7a-7c). As Rbp2 is known to bind to single-stranded RNA, specifically poly(U) and poly(G) RNA homopolymers (M. Sugita and Sugiura, 1994), and RNA-binding activity of Rbp2 appears to be required for it to execute clock function (Figure 6b), we sought to determine if an association between Rbp2 and KaiC could be detected in the presence of RNA. We were able to confirm that Rbp2 binds to poly(U) RNA (poly[rU]) (Figure 7d and 7e); however, an association between KaiC and Rbp2 was not observed after a 30 min incubation with purified KaiC, Rbp2, and poly(rU) (Figure 7e-7g). While it remains possible that Rbp2 and KaiC interact directly under conditions that were not tested, these data suggest that Rbp2 does not interact directly with KaiC and that other proteins are likely required to associate Rbp2 with KaiC.
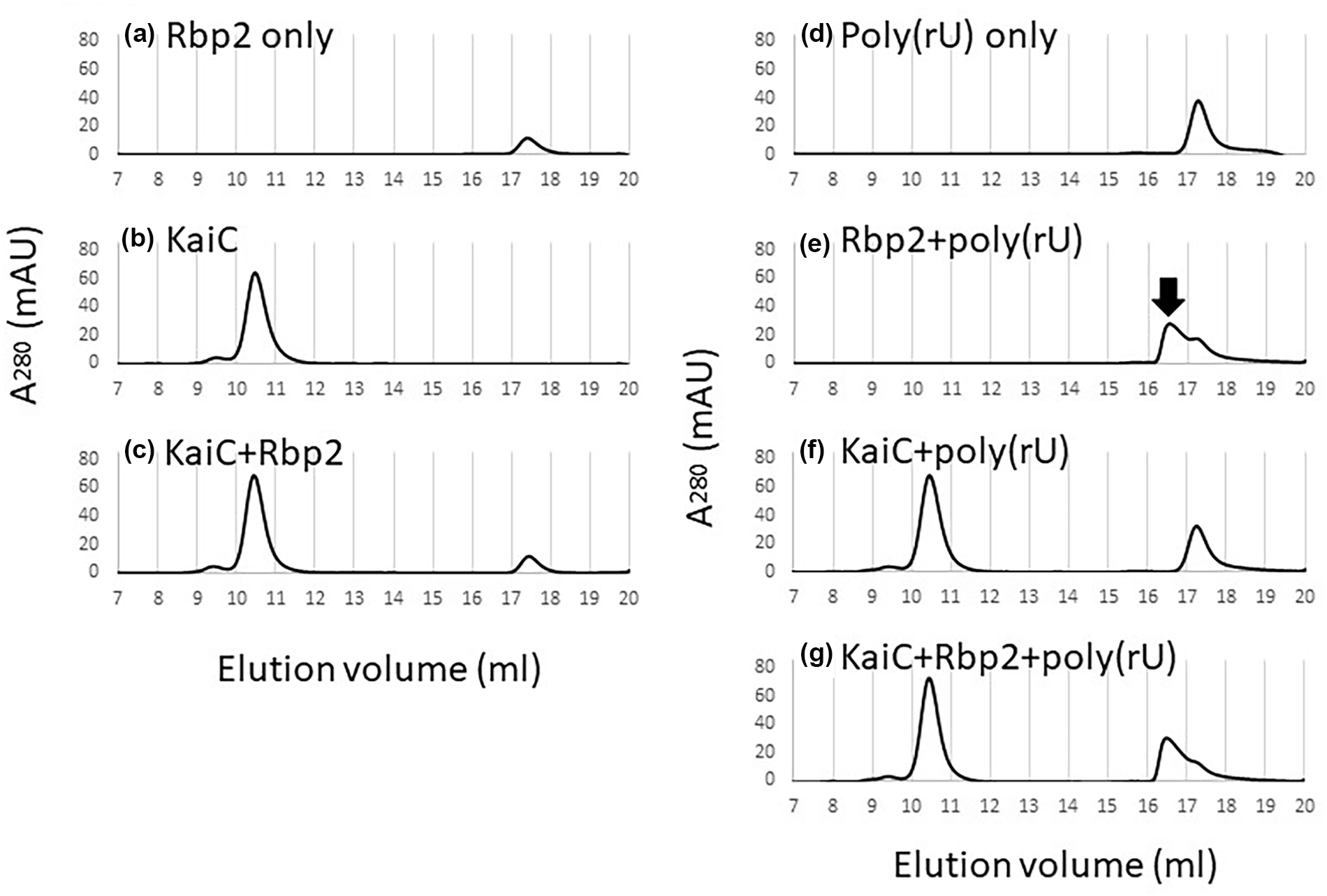
Lack of Rbp2 and KaiC complex formation in vitro suggests an indirect association. Chromatograms of gel filtration analysis showing the elution profiles of (a) Rbp2 only, (b) KaiC only, and (c) KaiC and Rbp2 together demonstrate an Rbp2-KaiC complex is not formed. Chromatograms of gel filtration analysis of (d) poly(rU) homopolymer only, (e) Rbp2 and poly(rU), (f) KaiC and poly(rU), or (g) KaiC and Rbp2 and poly(rU) suggest that the presence of RNA does not promote Rbp2-KaiC complex formation. However, a new peak that eluted earlier on the chromatogram (e), labeled with black arrow, was observed when Rbp2 was incubated with poly(rU), indicating the formation of a larger complex and confirming the ability of Rbp2 to bind RNA. Abbreviation: RNA = ribonucleic acid.
Discussion
Both the mechanism and function of KaiC temporal localization remain unclear although the discovery that Rbp2 affects this process provides new avenues for investigation. Because these two proteins do not show evidence of direct interaction in vitro, it is possible that Rbp2 bound to RNA creates a scaffold at or near the poles of cells, allowing for the docking or association of clock proteins. Such organization could potentially allow for clock complexes to associate at the appropriate stoichiometry and/or promote KaiC monomer shuffling, in which monomers are exchanged among KaiC hexamers. Monomer shuffling occurs during the dephosphorylation phase of the cycle and contributes to synchronization and stability of the Kai oscillator (Ito et al., 2007; Kageyama et al., 2006). Without this scaffold, it is possible that it would take longer for the complexes to assemble, leading to the observed long-period rhythms in gene expression.
We found that the CI domain of KaiC is sufficient to promote KaiC polar localization and that enhanced ATPase activity of KaiC correlates with enhanced polar localization. These data are consistent with the observations that ATPase activity of KaiC as well as its localization increase throughout the nighttime period (Cohen et al., 2014; Terauchi et al., 2007). Although the CI and CII domains are very similar, CI showed much higher propensity to localize. The hexamerization of CII is known to vary depending on phosphorylation state (Chang et al., 2011), which has not been characterized in vivo for the single domain. The recovery of CII polar localization upon overexpression may reflect a concentration-driven hexamerization that otherwise is unlikely to occur when the domain is expressed alone. Taken together, these data could suggest that the conformation of KaiC that promotes enhanced ATPase activity may also promote localization to the pole, perhaps by allowing for the binding of a partner that recruits KaiC to the pole.
We report that the ATPase activity of both KaiC and Rbp2 is involved in the localization of KaiC to one pole of the cell at night. Given the absence of a direct interaction between KaiC and Rbp2 in vitro, it is possible that KaiC ATPase and Rbp2 activities represent independent mechanisms that promote KaiC polar localization. However, it is also possible that they are connected. KaiC has enhanced ATPase activity when in association with KaiB and CikA (Mutoh et al., 2013; Tseng et al., 2017). It is possible that these proteins are responsible for associating with Rbp2, and we observe WT rhythmicity in vivo once this clock complex has been assembled.
Intriguingly, RRM-domain proteins similar to the one identified here have been reported to play roles in the circadian clock in eukaryotes. Clock-controlled RRM-containing proteins have been identified in the eukaryotic microalgae Gonyaulax and Chlamydomonas reinhardtii, as well as Neurospora, Arabidopsis, and Drosophila (Mittag, 2003; Schroeder et al., 2003; Staiger, 2001; Zhao et al., 2004). In Drosophila, the RRM-domain-containing protein LARK exhibits rhythms in abundance (McNeil et al., 1998), and its knockdown results in arrhythmic locomotor activity (Sundram et al., 2012). Moreover, abundance of the RNA-binding protein AtGRP7 of Arabidopsis thaliana oscillates with circadian rhythmicity, and overexpression represses these oscillations, suggesting it is part of a negative regulatory loop (Heintzen et al., 1997). The precise roles that these factors play in circadian function have not been determined, but they have all been proposed to function in circadian output and information relay from the oscillator to circadian controlled behaviors. While clock-controlled RRM-domain proteins have been implicated in circadian function in many different eukaryotic models, this report of RNA-binding activities or an RRM-domain protein in the prokaryotic clock model system suggests a novel conserved mechanism by which circadian clocks are regulated.
Supplemental Material
sj-docx-1-jbr-10.1177_07487304231188761 – Supplemental material for Roles for the Synechococcus elongatus RNA-Binding Protein Rbp2 in Regulating the Circadian Clock
Supplemental material, sj-docx-1-jbr-10.1177_07487304231188761 for Roles for the Synechococcus elongatus RNA-Binding Protein Rbp2 in Regulating the Circadian Clock by Briana M. McKnight, Shannon Kang, Tam H. Le, Mingxu Fang, Genelyn Carbonel, Esbeydi Rodriguez, Sutharsan Govindarajan, Nitsan Albocher-Kedem, Amanda L. Tran, Nicholas R. Duncan, Orna Amster-Choder, Susan S. Golden and Susan E. Cohen in Journal of Biological Rhythms
Footnotes
Acknowledgements
We thank Majid Ghassemian, director of the Biomolecular/Proteomics Mass Spectrometry Facility at UC San Diego, for his expertise and assistance with the collection and analysis of mass spectrometry data. This work was supported by NIH grant R35GM118290 to SSG and NSF CAREER Award MCB-1845953 to SEC.
Conflict Of Interest Statement
The authors have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplementary material is available for this article online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.