
Abstract
The aim of the present study was to develop a Polygenic Score–based model for molecular chronotype assessment. Questionnaire-based phenotypical chronotype assessment was used as a reference. In total, 54 extremely morning/morning (MM/M; 35 females, 39.7 ± 3.8 years) and 44 extremely evening/evening (EE/E; 20 females, 27.3 ± 7.7 years) individuals donated a buccal DNA sample for genotyping by sequencing of the entire genetic variability of 19 target genes known to be involved in circadian rhythmicity and/or sleep duration. Targeted genotyping was performed using the single primer enrichment technology and a specifically designed panel of 5526 primers. Among 2868 high-quality polymorphisms, a cross-validation approach lead to the identification of 83 chronotype predictive variants, including previously known and also novel chronotype-associated polymorphisms. A large (35 single-nucleotide polymorphisms [SNPs]) and also a small (13 SNPs) panel were obtained, both with an estimated predictive validity of approximately 80%. Potential mechanistic hypotheses for the role of some of the newly identified variants in modulating chronotype are formulated. Once validated in independent populations encompassing the whole range of chronotypes, the identified panels might become useful within the setting of both circadian public health initiatives and precision medicine.
Keywords
Chronotype is the most obvious behavioral expression of human endogenous circadian rhythmicity. It is the complex outcome of a number of molecular and physiological processes under circadian clock control. Nevertheless, from a behavioral standpoint, it can be easily described as the natural inclination to place one’s activity/sleep in different intervals of the 24-h day. Chronotype shows considerable variability in the population, ranging from early types, who are active in the morning hours and prefer to go to bed early, to late types, who find it more difficult to get up in the morning and easier to be active in the evening/early night hours. A significant proportion of the population has a less pronounced inclination (Horne and Ostberg, 1976; Roenneberg et al., 2003, 2007). Chronotype is often assessed on a phenotypic basis by use of self-administered questionnaires (Horne and Ostberg, 1976; Roenneberg et al., 2003; Ghotbi et al., 2020). The Horne-Östberg questionnaire, which explores a subject’s preference on when to perform specific activities over the course of the 24-h day (vide infra), is a good marker of chronotype when defined as diurnal preference (Vetter, 2018). The variable midsleep (i.e., the midpoint, expressed as clock time, between sleep onset and sleep offset) derived from the Munich Chronotype Questionnaire (Ghotbi et al., 2020; Roenneberg et al., 2003) is a good marker of chronotype defined as a proxy for the phase angle of entrainment (Vetter, 2018).
Twin and family studies suggest that approximately 50% of morning/evening preference can be explained by genetic factors (Klei et al., 2005; Koskenvuo et al., 2007; Barclay et al., 2010; Hsu et al., 2015). Nevertheless, the genetic bases of chronotype and its variability remain largely unknown. Only few genetic variations associated with chronotype have been investigated in depth, mainly because of their role in extreme sleep phenotypes such as advanced sleep-wake phase and delayed sleep-wake phase (Curtis et al., 2019; Ashbrook et al., 2020). For example, a mutation in the CASEIN KINASE 1E (CSNK1E) binding site of the PER2 gene (rs121908635; S662G) increases the stability of the protein resulting in an approximately 4 h phase advance (Toh et al., 2001) in individuals with familial advanced sleep-wake phase disorder. Interestingly, a similar phenotype results from a mutation (rs104894561; T44A) affecting the kinase activity of CASEIN KINASE 1D (CSNK1D; Xu et al., 2005). By contrast, a mutation in a splicing site in the CRY1 gene (rs184039278) results in the exon 11 skipping, which leads to a more persistent inhibitory activity of CRY1Δ within the negative feedback loop, at the core of the circadian clock, and therefore in a phase delay (Patke et al., 2017). However, less dramatic, non-pathological phenotypes have been associated with other genetic variations, the contribution of which is minor, less consistent, and often questioned. For example, a variable number tandem repeat (VNTR) polymorphism in exon 18 of the PER3 gene (rs57875989), with 2 alleles encoding four or five 18 aa repeats, has also been associated with chronotype. The longer allele (PER5) has been associated with morningness, while the shorter (PER4) with eveningness (Archer et al., 2003). Since each repeat contains several predicted CSNK1D phosphorylation sites, it has been proposed that the number of repeats could affect the phosphorylation state of the protein, with potential effects on its stability (Archer et al., 2003). Interestingly, this VNTR polymorphism has also been implicated in light sensitivity, with the 2 variants differently affecting melatonin suppression (Chellappa et al., 2012). More recently, we have simultaneously assessed the PER3 VNTR polymorphism and a missense single-nucleotide polymorphism (SNP) (rs228697) in the coding region of the same gene (Turco et al., 2017). This single-nucleotide variation results in an amino acid substitution (P864A) altering the secondary structure of a conserved domain relevant to the binding with NCK, an adaptor protein involved in the interaction between PER3 and CSNK1 (Akashi et al., 2002; Lussier and Larose, 1997; Turco et al., 2017). The PER3G variant showed a significant association with morningness, both as a stand-alone and in combination with the PER34 allele (Turco et al., 2017). All these studies suggest that chronotype is a complex trait—which is also supported by its being normally distributed (Ashbrook et al., 2020)—the definition of which is likely to benefit from a genome-wide approach.
The first 2 genome-wide association studies (GWAS; Gottlieb et al., 2007; Hu et al., 2016) have shown that most chronotype-associated loci are in close proximity to genes involved in circadian and sleep regulation. A recent meta-analysis of nearly 700,000 individuals (from the 23andMe Inc. data set, https://research.23andme.com/; and the UK Biobank, https://www.ukbiobank.ac.uk/) has identified 351 genomic loci associated with chronotype (Jones et al., 2016, 2019) including SNPs on some of the main circadian clock genes (PER1-3, CRY1, FBXL3, and ARNTL).
In all these GWAS studies, chronotype was self-reported and genotyping performed by custom high-throughput platforms which cannot detect rare SNPs, length polymorphisms, and short insertions or deletions (Höglund et al., 2019). Nevertheless, these could represent a valuable source of information to identify additional chronotype contributors, as was the case for the PER3 VNTR (Archer et al., 2003; Turco et al., 2017).
Our aim was to develop a Polygenic Score–based model for chronotype assessment based on genotyping by sequencing of the entire genetic variability of 19 target genes known to be involved in circadian rhythmicity and sleep duration, only in extremely morning/morning (MM/M) and extremely evening/evening (EE/E) individuals. This strategy has already been proven to increase the probability of finding novel genetic variants affecting complex continuous phenotypes by sequencing relatively small samples (Amanat et al., 2020).
Materials and Methods
A schematic overview of the experimental workflow is shown in Supplementary Figure S1.
Study Population
Between 2012 and 2019, a cohort of 679 healthy individuals were recruited during University of Padova popular science events (Turco et al., 2017) and educational workshops. Participants were asked to donate a buccal DNA sample and to complete a comprehensive sleep-wake assessment (Turco et al., 2017).
Chronotype Assessment
Chronotype was assessed by the self-administered Morningness-Eveningness Questionnaire (Horne and Ostberg, 1976; Tonetti and Natale, 2019), based on a set of questions asking the subject to indicate when, in specific periods of the 24-h day, they would prefer to place specific activities, if they were completely free to plan their day. Total scores range from 16 to 86: scores ≤ 41 define E types, with EE ≤ 30; scores ≥ 59 define M types, with MM ≥ 70; and scores between 42 and 58 define intermediate types. In our study population, 153 participants showed MM/M (22.5%) and 152 EE/E (22%). Analysis of variance (ANOVA; post hoc Tukey test) was used to compare age among groups.
Sample Collection, Processing, and Selection
Buccal DNA samples were self-collected by brushing the inside of the cheeks with a swab for about 15 sec. Samples were stored at room temperature, and DNA extracted within 12 h by the BuccalAmp QuickExtract kit (Epicentre, illumina.com) according to the manufacturer’s instructions. DNA was further purified by phenol/chloroform extraction (one volume of phenol:chloroform:isoamyl alcohol [25:24:1] and two volumes of EtOH 100%), and resuspended in water. DNA amount and integrity were assessed by NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific; thermofisher.com) and agarose gel electrophoresis, respectively. Then, selected DNA samples were further quantified with the QuantiFluor dsDNA System (Promega; promega.com) to avoid sample overestimation due to RNA contamination. Among the 305 participants who provided a DNA sample and showed MM/M or EE/E, 96 DNA samples were selected for genotyping on the basis of DNA integrity and concentration. These included 10 MM, 42 M, 30 E, and 14 EE types (Table 1). Expected differences in age and sex were observed, with MM/M types being older (significant difference) and more frequently women (non-significant difference) than EE/E types (Table 1; Adan and Natale, 2002; Robilliard et al., 2002; Turco et al., 2017).
Characteristics of the study population (only the DNA samples selected for genotyping).

p < 0.001 significance of the difference between extremely evening and extremely morning individuals.
p < 0.05 significance of the difference between evening and extremely morning individuals.
Abbreviations: MEQ = Morningness-Eveningness Questionnaire.
Target Selection
The entire genetic variability of 19 target genes, involved in circadian rhythmicity and sleep duration, was assessed. Among the main core components of the endogenous clock, 14 circadian genes, which have previously been associated with chronotype in candidate gene studies (Mishima et al., 2005; Carpen et al., 2006; Matsuo et al., 2007; He et al., 2009; Lee et al., 2011; Etain et al., 2014; Hida et al., 2014; Parsons et al., 2014; Dmitrzak-Węglarz et al., 2016; Hirano et al., 2016; Jankowski and Dmitrzak-Weglarz, 2017; Patke et al., 2017; Turco et al., 2017; Kurien et al., 2019) and genome-wide analyses (Gottlieb et al., 2007; Hu et al., 2016; Jones et al., 2016, 2019; Lane et al., 2016; Maukonen et al., 2020), were selected: PER1, PER2, PER3, CRY1, CRY2, ARNTL, CLOCK, TIMELESS, NPAS2, NCK, RORA, NR1D1, NR1D2, and CSNK1E. A list of the most relevant SNPs on clock genes which have previously been associated with chronotype is provided in Supplementary Table S1. In addition, 5 genes, known to play a significant role in sleep duration (Zhang and Fu, 2020), were also included: ABCC9, BHLHE41, DRD2, ADA, and FABP7.
S.P.E.T. Sequencing Strategy and Primer Design
Targeted genotyping by sequencing was performed using the Single Primer Enrichment Technology (S.P.E.T.; Barchi et al., 2019) based on the Illumina sequencing platform (illumina.com). A single primer is designed for the genotype calling of one selected SNP by sequencing a small region of about 150 bp around the variant site. Thus, a panel of thousands of primers allows genotype calling of thousands of SNPs at the same time. Furthermore, the technology allows the targeted sequencing of a gene by simply increasing the number of primers up to complete coverage. A specific panel of primers was designed by Tecan Genomics (lifesciences.tecan.com) on the sequence of the 19 target genes including exons, regulatory sequences up to 1000 bp upstream the transcription start site, and intronic regions 200 bp over the splicing site, to ensure coverage of exon peripheral regions and to include the canonical splice site consensus sequences such as branchpoints and B-boxes (Mercer et al., 2015). Primers were also designed to specifically assess 18 SNPs previously associated with chronotype but located far from the selected target regions (a complete list is available in Supplementary Table S2). The resulting 5526 primers (a complete list is available in Supplementary Table S3) were aligned to the reference genome (Homo sapiens, hg38, UCSC iGenomes) to verify the complete coverage of all the genomic regions of interest using the custom track function on the UCSC Genome Browser (genome.ucsc.edu).
Genotyping and Quality Control
Targeted genotyping by sequencing and quality controls (QCs) were performed by IGA Technology Services (igatechnology.com; Udine, Italy). Libraries were prepared using 20 ng/µL of DNA as input according to the Allegro Targeted Genotyping protocol (NuGEN Technologies; nugen.com). Libraries were quantified using the Qubit 2.0 Fluorometer (Thermo Fisher Scientific; thermofisher.com) and their size distribution checked by the Bioanalyzer High Sensitivity DNA assay (Agilent technologies; agilent.com). Diluted libraries were quantified through quantitative PCR (qPCR) using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad; bio-rad.com) and run on the Illumina NovaSeq 6000 (Illumina; illumina.com) with 2 × 150 paired-end reads. Base calling and demultiplexing were performed using bcl2fastq v2.20 (Illumina; illumina.com). Raw reads quality assessment and adapter trimming were performed using Cutadapt (Martin, 2011) with default parameters. In further detail, not only the adapters but also the first 40 bp of each paired-end read (R1) were trimmed to exclude the primer sequence from the SNP calling. Reads alignment to the reference genome (Homo sapiens, hg38, UCSC iGenomes) and the selection of uniquely aligned reads (mapping quality > 10) were performed using ERNE (Del Fabbro et al., 2013) v1.4.6 and BWA-MEM (Li and Durbin, 2009) v0.7.17 with default parameters. SNP calling was performed using gatk-4.0 following the software best practice for germline short-variant discovery proposed by DePristo et al. (2011): (1) per-sample variants calling on target regions using HaplotypeCaller with default parameters; (2) multiple samples consolidation using GenomicsDBImport with default parameters; (3) joint genotyping using GenotypeGVCFs with default parameters; (4) selection of SNPs using SelectVariants and quality filtering of SNPs using VariantFiltration (filter expression used: Quality by Depth [QD] < 2.0, Mapping Quality [MQ] < 40.0, and MQRankSum < -12.5). SNPs were annotated according to the Single Nucleotide Polymorphism Database (dbSNP) b155 v2 (https://www.ncbi.nlm.nih.gov/snp/).
Genotyping Data Analysis
Quality control and association analyses were conducted with PLINK, versions 1.9 and 2.0 (Chang et al., 2015). The QC pipeline proposed by Marees et al. (2018) was applied on genotype data prior to conducting association analysis. In particular, results were considered acceptable if missingness of SNPs was < 10%, missingness of SNPs per individual was < 10%, minor allele frequency (MAF) was ≥1%, deviation from Hardy-Weinberg equilibrium had a significance of p>1e-10, and heterozygosity was within 3 SDs from mean heterozygosity. There were no related individuals among the 96 donors of the genotyped DNA samples. Finally, a multidimensional scaling analysis (MDS) was performed on the total number of high-quality SNPs to check for population stratification.
Polygenetic Risk Score Calculation
Association analysis was conducted by binary logistic regression (MM/M versus EE/E types) and adjusted (additive model) for sex, age, and the first 2 principal components of the MDS analysis. False discovery rate was calculated by Benjamini-Hochberg multiple testing correction (Benjamini and Hochberg, 1995). The individual genetic predisposition for morningness/eveningness was evaluated by Polygenetic Score (PS) analysis using PRSice-2 (Euesden et al., 2015). Significantly associated SNPs were linkage disequilibrium (LD)-clumped with an LD threshold (R2) = 0.5, and a range of 50 kb to reveal independent chronotype-associated SNPs (lead SNPs). To prevent overfitting, the proportion of variance explained by our data set was estimated as the average partial pseudo R2 (Nagelkerke’s R2 for binary chronotype; Nagelkerke, 1991) of the best models generated in a 10 × 5-fold cross-validation with a maximum p-value threshold of 0.05. A cross-validation approach (Manor and Segal, 2013) was used to obtain a variant ranking based on their predictive performance. In particular, lead SNPs were ranked based on the number of best models in which they were included. If 2 or more SNPs shared the same position, they were ordered based on their average association p value in the 10 × 5-fold cross-validation. The proportion of variance explained by different SNP sets was estimated as the average partial pseudo R2 of the models generated in a new 10 × 5-fold cross-validation using the PRSset function in PRSice. The proportion of variance explained by the PS-based model is intended as an estimate of the predictive value of the model.
Results
Primer design resulted in 5526 primers (Supplementary Figure S2 shows how these were distributed within the Per3 gene, as a representative example). Targeted sequencing generated about 28 million paired-end reads with an average of 290,649 reads per sample and a mapping rate of 98% (reference genome: homo_sapiens_hg38-iGenomes).
Variant calling identified 25,030 variants including not only SNPs but also indels, polymorphic repetitive elements, and false positives. Thus, variants needed to be quality filtered. By applying Quality by Depth and Mapping Quality stringent criteria, 8968 high-quality SNPs were identified with a 95.4% total genotyping rate. QC resulted in a final sample of 93 individuals (50 MM/M, 43 EE/E) and 2868 variants, with an average call rate of 99%. MDS analysis revealed no significant population stratification with a maximum proportion of variance explained by a principal component of 4.97%. However, the first 2 components of the MDS analysis were included in the association model as covariates, in addition to sex and age, to account for a possible minor source of distortion. Chronotype (MM/M versus EE/E) association p value and chromosomal position of each variant were used in the LD-clumping procedure to remove 1365 correlated variants and retain the 1503 independent association signals (lead variants; Suppl. Table S4). Only 2 significant associations (rs3213578 and rs228699, 2 PER3 intron variants; Table 2) were detected when p values were corrected for multiple testing. However, higher p values did not impinge on the development of a predictive model based on clumping and thresholding methods, where the association p values are used to establish a ranking of the variants but their significance level is not relevant.
List of the 35 predictive variants (complete panel).
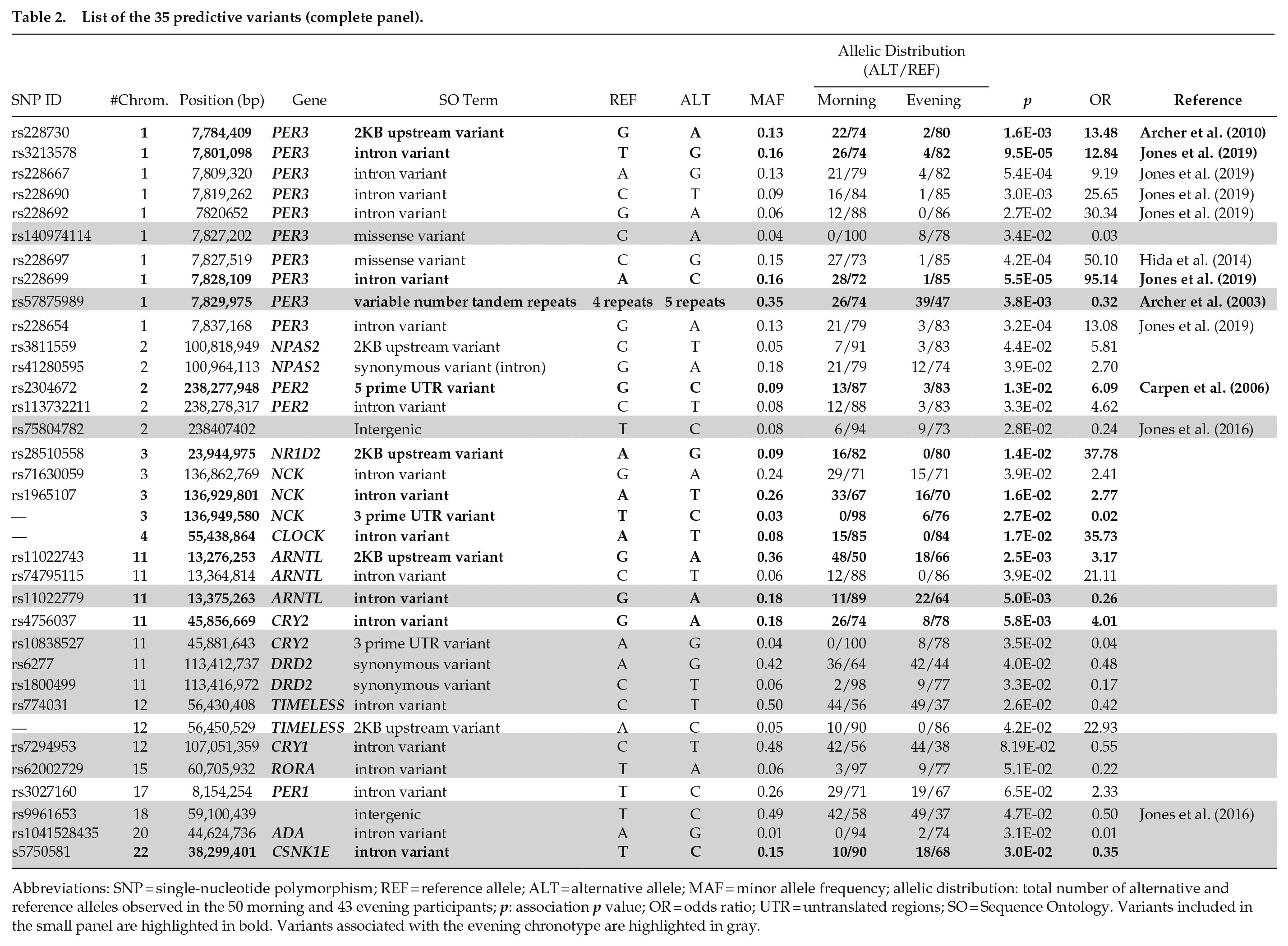
Abbreviations: SNP = single-nucleotide polymorphism; REF = reference allele; ALT = alternative allele; MAF = minor allele frequency; allelic distribution: total number of alternative and reference alleles observed in the 50 morning and 43 evening participants; p: association p value; OR = odds ratio; UTR = untranslated regions; SO = Sequence Ontology. Variants included in the small panel are highlighted in bold. Variants associated with the evening chronotype are highlighted in gray.
To obtain a ranking of the most predictive variants, the data set was repeatedly resampled in a 10 × 5-fold cross-validation, a PS-based model was generated for each resampling, and then the final ranking was determined based on the number of models in which each variant was included. This way, priority was given to the most informative variants, showing both high association and high MAF. For the PS modeling step, a p-value threshold of 0.05 was used to avoid inflation. A total of 83 lead significant variants were ranked (Suppl. Table S4). The predictive performance of 83 sets, encompassing the first N (1 ≤ N ≤ 83) variants, was assessed in a new 10 × 5-fold cross-validation. The first 13 variants (from here onward “small panel”) explained more than 80% of the variance of the population (Figure 1a), a result which was comparable with that of the predictive model with no variants selection (78.9%; Figure 1a). A second peak in explained variance (81.0%; Figure 1a) was reached with the first 35 variants (from here onward “large panel”). A further increase in the number of included variants did not improve the performance of the predictive model to any significant extent. PS based on both the large and the small panels was then calculated for all 96 genotyped subjects. Both PS distributions (Figure 1b) successfully separated EE/E from MM/M types (p < 10−14).
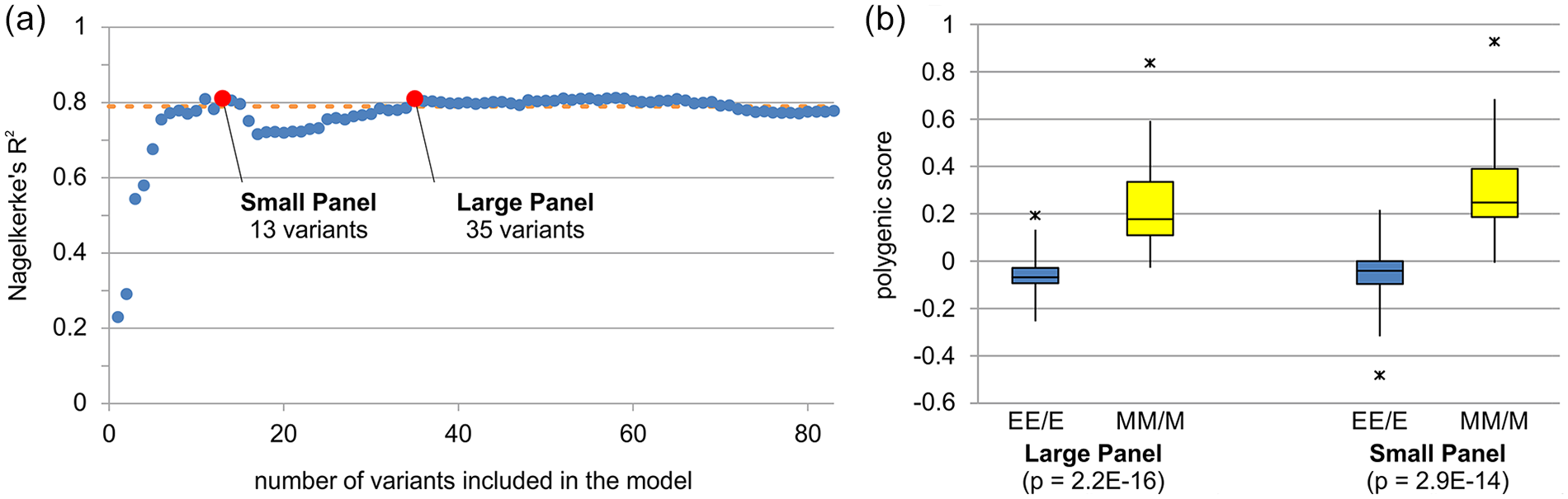
Polygenic score analysis. (a) Estimated variance explained by the model as the number of included variants increases. The proportion of explained variance is reported as the average partial pseudo Nagelkerke’s R2 in a 10 × 5-fold cross-validation. Red dots represent the variance explained by the first 13 (small panel) and 35 variants (large panel), respectively. The orange dotted line represents the variance explained by the predictive model without variants selection. (b) Differences between polygenic score distributions in the 44 evening (EE/E; blue) and 52 morning (MM/M; yellow) participants (median; upper and lower quartile; minimum and maximum) for the large (left) and the small (right) panels; p values refer to Mann-Whitney U tests. Asterisks mark potential outliers, that is, observations with a standardized value >3 or <−3. Abbreviations: EE/E extremely evening/evening; MM/M extremely morning/morning. Color version of the figure is available online.
The 35 highly predictive variants of the large panel were distributed on 15 out of the 19 target genes (Table 2). No predictive SNPs were found on the circadian gene NR1D1 or on 3 out of the 5 genes involved in sleep duration (ABCC9, BHLHE41, and FABP7). Of the 35 variants, 12 have been previously associated with chronotype (Table 2; Suppl. Table S1), including 2 SNPs (rs75804782 and rs9961653) on intergenic regions belonging to the group of 18 variants that we included even though they were located far from the target genes (Suppl. Table S2).
Ten of the highly predictive variants mapped on PER3, 3 on ARNTL, and 3 on NCK. The alternative alleles (i.e., alternative to the “wild-type” allele more commonly observed in the reference population) of 21 SNPs were associated with the MM/M chronotype (Table 2; OR > 1), while the alternative alleles of the remaining 14 variants were associated with the evening chronotype (Table 2; OR < 1). The 13 highly predictive variants of the small panel (Table 2) mapped on 8 clock genes (ARNTL, CLOCK, CRY2, CSNK1E, NCK, NR1D2, PER2, and PER3), with 9 and 4 being variants associated with MM/M and EE/E chronotype, respectively.
Discussion
In this study, we defined a panel of 35 predictive genetic polymorphisms for potential use in chronotype assessment. In further detail, we identified 23 novel variants which, in combination with 12 SNPs previously associated with chronotype as stand-alone, led to the development of a PS-based model with an estimated predictive value of about 80% in our population. Our findings suggest that the development of a predictive model for a complex trait, such as chronotype, can benefit from complete assessment of the genetic variability of a number of selected target genes. Our experimental design confirmed findings from previous GWAS and candidate gene studies, and also led to the identification of a non-negligible number of novel SNPs.
Our results confirmed the key role of PER3 in chronotype modulation, with the highest number of our identified highly predictive variants (10 out of 35) mapping on its gene. Unlike PER1 and PER2, which are essential for circadian rhythms’ maintenance and light responses in the master clock, PER3 has long been considered to exert its timekeeping function mostly in specific peripheral clocks (Pendergast et al., 2010, 2012). More recently, a role for PER3 in stabilizing PER1/PER2 has been proposed (Zhang et al., 2016). This hypothesis is supported by the high number of association studies linking human PER3 polymorphisms and chronotype (Hida et al., 2014; Parsons et al., 2014; Turco et al., 2017; Jones et al., 2019), sleep (Hasan et al., 2014), and mood (Zhang et al., 2016). rs228697 and rs140974114 are the only missense variants included in the model and are both located on the PER3 gene. rs228697 has been already associated with chronotype in previous studies (Hida et al., 2014; Turco et al., 2017) and it is thought to modify the secondary structure of the protein and its phosphorylation dynamics, affecting the interaction with the scaffold protein NCK, and PER3 stability.
Further analysis, either molecular or in silico, of the effects of the novel variants included in the predictive model could provide valuable insights into the molecular mechanisms underlying chronotype. For example, rs140974114 has never been associated with chronotype before. The alternative allele is associated with eveningness and leads to the substitution of a serine with an asparagine (S750N) in the PER3 protein (Figure 2). The polymorphism lies within the CSNK1E binding domain (555-760 aa; UniProt.org), close to a nuclear localization signal (NLS) motif (729-745 aa; UniProt.org), and the amino acid substitution could affect a putative phosphorylation site (phosphonet.ca). The phosphorylation status of PER proteins is known to influence stability and nuclear import, and more specifically, phosphorylation sites are required for optimal CSNK1E-mediated nuclear import of PER3 in mice (Akashi et al., 2002). In humans, the role of CSNK1E is less understood. However, the obliteration of the putative phosphorylation site in the S750N variant might alter the masking/unmasking of the NLS, resulting in different kinetics of the nuclear translocation of PER3.

Schematic representation of the hypothetical effect of the S758N mutation on the CSNK1E-binding domain of PER3. The blue bar represents the CSNK1E-binding domain on the PER3 protein. The NLS is represented as a green box. Putative phosphorylation sites are highlighted in red. The red arrow indicates the mutation site and the resulting amino acid substitution. Abbreviation: NLS = nuclear localization signal. Color version of the figure is available online.
Among the most relevant genetic variations, we also found the PER3 VNTR, which, despite a low significance association with chronotype, was included in the small panel. The previously reported synergistic effect on chronotype of PER3 VNTR and the P864A substitution (rs228697; Turco et al., 2017) was confirmed also in this series.
Seven variants, which could alter gene expression, were also identified. Three were synonymous variants, two on the DRD2 gene, and another one in NPAS2, possibly affecting transcription, splicing, mRNA stability, and/or co-translational folding (Zeng and Bromberg, 2019). Four were variants located in the putative promoter regions of NR1D2, ARNTL, TIMELESS, NCK, and PER3, possibly altering gene expression by impairing a transcription factor binding site. For example, the rs228730 SNP on PER3 promoter region falls in a CpG site between 2 Sp1 (specificity protein 1) transcription factor binding sites and its influence on PER3 expression has been experimentally confirmed (Archer et al., 2010). Finally, 4 additional variants fell on the untranslated regions (UTR) of NCK, CRY2, and PER2. Mutations on these regions could affect mRNA stability, localization, and translation through effects on the specific binding sites of regulatory proteins and non-coding RNAs (Steri et al., 2018). For example, an SNP on the 3′UTR of the NCK gene (Ch12:136949580T>C), together with other 2 correlated SNPs (Ch12:136949584G>A and Ch12:136949587G>T), disrupts the hsa-miR-6875-3p recognition element site (predicted by miRDB, Target score 98; mirdb.org). Apparently, none of the intron variants fell on a canonical splice site (branchpoints or B-boxes), but their role in affecting alternative splicing through splicing regulatory elements (intronic splicing enhancer or silencer) cannot be excluded.
The estimated proportion of variance explained by our genetic model (81%) is above the previously estimated heritable component of chronotype (up to 52%; Klei et al., 2005; Koskenvuo et al., 2007; Barclay et al., 2010; Hsu et al., 2015). This is most likely related to the fact that, in this instance, intermediate types were not included. Our model (both the large and the small panels) will need validation in independent samples encompassing the whole range of chronotypes. Larger samples will also allow a linear regression approach based on the 5 chronotype classes (MM, M, intermediate, E, EE) or the continuous Morningness-Eveningness Questionnaire score.
Our study has a number of limitations, including the inherent confounding associated with phenotyping for sleep-wake features in a real-life environment, the relatively small sample size, and the exclusion of intermediate types.
We plan to make use of this model to develop a multiplex genotyping assay to define chronotype by a molecular approach, which may have advantages in settings where questionnaire-based chronotype assessment is confounded, for example, by disease or hospitalization (Bano et al., 2014). Furthermore, the relatively small number of predictive variants included in the model will allow development of a faster and accessible PCR-based genotyping assay to replace the high-throughput approach. The use for this can be envisaged in both public health initiatives and in precision medicine. Possibly even more importantly, one can imagine a process whereby further studies are performed to validate the model and the model is also progressively refined encompassing newly identified loci. This process is likely to lead to future, hypothesis-driven research.
In conclusion, two useful panels for the molecular assessment of chronotype were developed based on an innovative genotyping-by-sequencing approach of 19 clock and sleep-related genes in a sample of selected individuals with opposite chronotypes.
Supplemental Material
sj-tif-1-jbr-10.1177_07487304221099365 – Supplemental material for Toward a Molecular Approach to Chronotype Assessment
Supplemental material, sj-tif-1-jbr-10.1177_07487304221099365 for Toward a Molecular Approach to Chronotype Assessment by Alberto Biscontin, Lisa Zarantonello, Antonella Russo, Rodolfo Costa and Sara Montagnese in Journal of Biological Rhythms
Supplemental Material
sj-tif-2-jbr-10.1177_07487304221099365 – Supplemental material for Toward a Molecular Approach to Chronotype Assessment
Supplemental material, sj-tif-2-jbr-10.1177_07487304221099365 for Toward a Molecular Approach to Chronotype Assessment by Alberto Biscontin, Lisa Zarantonello, Antonella Russo, Rodolfo Costa and Sara Montagnese in Journal of Biological Rhythms
Supplemental Material
sj-xlsx-3-jbr-10.1177_07487304221099365 – Supplemental material for Toward a Molecular Approach to Chronotype Assessment
Supplemental material, sj-xlsx-3-jbr-10.1177_07487304221099365 for Toward a Molecular Approach to Chronotype Assessment by Alberto Biscontin, Lisa Zarantonello, Antonella Russo, Rodolfo Costa and Sara Montagnese in Journal of Biological Rhythms
Supplemental Material
sj-xlsx-4-jbr-10.1177_07487304221099365 – Supplemental material for Toward a Molecular Approach to Chronotype Assessment
Supplemental material, sj-xlsx-4-jbr-10.1177_07487304221099365 for Toward a Molecular Approach to Chronotype Assessment by Alberto Biscontin, Lisa Zarantonello, Antonella Russo, Rodolfo Costa and Sara Montagnese in Journal of Biological Rhythms
Supplemental Material
sj-xlsx-5-jbr-10.1177_07487304221099365 – Supplemental material for Toward a Molecular Approach to Chronotype Assessment
Supplemental material, sj-xlsx-5-jbr-10.1177_07487304221099365 for Toward a Molecular Approach to Chronotype Assessment by Alberto Biscontin, Lisa Zarantonello, Antonella Russo, Rodolfo Costa and Sara Montagnese in Journal of Biological Rhythms
Supplemental Material
sj-xlsx-6-jbr-10.1177_07487304221099365 – Supplemental material for Toward a Molecular Approach to Chronotype Assessment
Supplemental material, sj-xlsx-6-jbr-10.1177_07487304221099365 for Toward a Molecular Approach to Chronotype Assessment by Alberto Biscontin, Lisa Zarantonello, Antonella Russo, Rodolfo Costa and Sara Montagnese in Journal of Biological Rhythms
Footnotes
Acknowledgements
The study and authors AB and LZ were supported by a Supporting TAlent in ReSearch@University of Padova STARS@UNIPD 2019 “Consolidator Grants (STARS-CoG)” to author SM. The study was also supported by the Comparative Insect Chronobiology (CINCHRON), EU Horizon 2020, Marie Sklodowska-Curie Initial Training Network (grant agreement N° 765937) to author RC.
Conflict of Interest Statement
The author(s) have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.