
Abstract
Over 90% of neurons within the suprachiasmatic nucleus (SCN) express γ-aminobutyric acid (GABA). Although GABA is primarily an inhibitory neurotransmitter, in vitro studies suggest that the activation of GABAA receptors (GABAAR) elicits excitation in the adult SCN. The ratio of excitatory to inhibitory responses to GABA depends on the balance of chloride influx by Na+-K+-Cl– cotransporter 1 (NKCC1) and chloride efflux by K+-Cl– cotransporters (KCCs). Excitatory responses to GABA can be blocked by inhibition of the inward chloride cotransporter, NKCC1, with the loop diuretic bumetanide. Here we investigated the role of NKCC1 activity in phase shifting the circadian pacemaker in response to photic and nonphotic signals in male Syrian hamsters housed in constant darkness. In the early subjective night (CT 13.5), injection of bumetanide into the SCN reduced light-induced phase delays. However, during the late subjective night (CT 19), bumetanide administration did not alter light-induced phase advances. Injection of bumetanide during the subjective day (CT 6) did not alter the phase of free-running circadian rhythms but attenuated phase advances induced by injection of the GABAAR agonist muscimol into the SCN. These data support the hypothesis that the excitatory effects of endogenously released GABA contribute to the ability of light to induce phase delays, thereby contributing to the most important function of the circadian system, its entrainment with the day-night cycle. Further, the finding that bumetanide inhibits the phase-advancing effects of muscimol during the subjective day supports the hypothesis that the excitatory responses to GABA also contribute to the ability of nonphotic stimuli to phase shift the circadian pacemaker.
The suprachiasmatic nucleus (SCN) plays a key role in the circadian control of physiology and behavior in mammals, including humans (Moore and Eichler, 1972; Stephan and Zucker, 1972; Cohen and Albers, 1991). Two of the most important functions of the SCN are generating self-sustained oscillations and adjusting the period of those oscillations with the 24-h day-night cycle through the process of entrainment (Daan and Pittendrigh, 1976; Johnson et al., 2003). Light in the early subjective night phase delays the pacemaker; light in the late subjective night phase advances the pacemaker; while light in the subjective day does little to shift the clock (Decoursey, 1960; Daan and Pittendrigh, 1976). Light is transduced in specialized retinal ganglion cells and communicated to the SCN via the release of glutamate from terminals of the retinohypothalamic tract (RHT) (Hendrickson et al., 1972; Moore and Lenn, 1972; Pickard, 1982; Colwell et al., 1991; Mintz et al., 1999; Berson et al., 2002; Sollars et al., 2003).
Although the RHT appears to be the major light input pathway regulating many of the effects of the strongest zeitgeber on the SCN, other stimuli can also induce phase shifts of the pacemaker. Nonphotic phase shifting stimuli include the injection of neuropeptide Y-like peptides, discrete bouts of resident-intruder interactions, or exposure to a novel running wheel (Albers and Ferris, 1984; Mistlberger, 1991; Biello and Mrosovsky, 1995; Biello and Mrosovsky, 1996; Mistlberger et al., 2003). In nocturnal rodents, nonphotic stimuli produce a pattern of phase shifts (i.e., small phase delays during subjective night and large phase advances during subjective day) that differ greatly from those produced by light. Stimuli that induce nonphotic phase shifts are relayed to the SCN via projections from other brain regions including a direct projection from the intergeniculate leaflet and a serotonin-containing projection from the raphe (Swanson et al., 1974; Ribak and Peters, 1975; Meyer-Bernstein and Morin, 1996; Morin, 1999; Morin, 2013). Finally, the SCN provides output signals that serve to entrain physiological systems throughout the body with the day-night cycle (Kalsbeek et al., 2006; Morin, 2013).
The SCN is a network of γ-amino butyric acid (GABA)-containing cells. GABA synthesis and transporter proteins as well as GABA receptors are expressed in more than 90% of SCN neurons (van den Pol and Tsujimoto, 1985; van den Pol and Gorcs, 1986; Okamura et al., 1989; Moore and Speh, 1993; O’Hara et al., 1995; Castel and Morris, 2000; Albers et al., 2017). Two major classes of GABA receptors, GABAA and GABAB receptors (GABAARs and GABABRs, respectively), mediate the effects of GABA within the SCN. GABAARs are ligand-gated chloride ion channels whose subunit composition determines their pharmacological properties, subcellular localization, and rhythmic regulation within the SCN (Farrant and Nusser, 2005; Walton et al., 2017). GABABRs are metabotropic G-protein-coupled receptors that can be found on presynaptic, postsynaptic, and extrasynaptic membranes (Enna and McCarson, 2013). GABA can modulate the ability of the pacemaker to phase shift in response to light by acting on GABAARs and GABABRs (Ralph and Menaker, 1985; Ralph and Menaker, 1986; Ralph and Menaker, 1989; Gribkoff et al., 1998; Mintz et al., 2002; Albers et al., 2017). The GABAAR agonist muscimol and the GABABR agonist baclofen administered into the SCN inhibit phase delays in response to light in the early subjective night and decrease light-induced phase advances in the late subjective night (Gillespie et al., 1997; Gillespie et al., 1999; Novak and Albers, 2004). In addition, GABAAR and GABABR antagonists administered in the SCN enhance phase delays to light in the early subjective night (Gillespie et al., 1996). GABAARs also play a critical role in mediating the phase-delaying effects of light. The sustained activation of GABAARs (>4 h) is both necessary and sufficient to regulate the phase-delaying effects of light in the early subjective night (Hummer et al., 2015).
GABAAR signaling is also critical in SCN processing of nonphotic phase shifting stimuli (Webb et al., 2014). For example, bicuculline blocks neuropeptide Y (NPY)-induced phase advances when given in the SCN during the middle of the subjective day (Huhman et al., 1995; Huhman et al., 1997; Gribkoff et al., 1998). Interestingly, muscimol and diazepam, drugs that act on GABAARs, phase advance the pacemaker when administered during the subjective day in nocturnal rodents but induce phase delays in the diurnal grass rat (Smith et al., 1989; Mintz et al., 2002; Novak and Albers, 2004; McElroy et al., 2009). Thus, activation of GABAARs plays a key role in regulating SCN mechanisms mediating the phase shifting effects of both photic and nonphotic stimuli.
Although GABA is most commonly viewed as the primary inhibitory neurotransmitter in the adult brain, it can also have excitatory effects. The actions of cation chloride cotransporters (CCCs) are critical in determining whether GABA is hyperpolarizing or depolarizing (Xu et al., 1994; Gillen et al., 1996; Payne et al., 1996; Markadieu and Delpire, 2014). Two main types of CCCs regulate intracellular chloride. Na-K-2Cl cotransporters (NKCCs) transport chloride ions (stoichiometry 1Na+: 1K+: 2Cl–) into cells in 2 identified isoforms, NKCC1 and NKCC2. K-Cl cotransporters (KCCs) extrude chloride ions from the cell (stoichiometry 1K+: 1Cl–), with 4 identified isoforms (KCC1-4). Thus, as the GABAAR opens a chloride pore when active, the probability of depolarizing a GABAAR-expressing neuron is determined by the ratio of the activity of chloride influx (NKCC1) to chloride efflux (KCCs) (for review, see Markadieu and Delpire, 2014).
The cellular responses to GABA within the SCN have been extensively studied using a variety of approaches, and yet there is little consensus on when, where, and whether GABA produces excitatory responses in SCN neurons (Wagner et al., 1997; Liu and Reppert, 2000; De Jeu and Pennartz, 2002; Itri et al., 2004; Albus et al., 2005; Aton et al., 2006; Choi et al., 2008; Irwin and Allen, 2009; Myung et al., 2012; Freeman et al., 2013; Farajnia et al., 2014; Albers et al., 2017). While there is agreement that GABA has substantial inhibitory effects in the SCN, some studies report that GABA is exclusively inhibitory, others that GABA can be excitatory during the day, and still others that GABA can be excitatory during the night. There is also controversy on whether the excitatory effects of GABA are more frequently observed in different subregions of the SCN. Importantly, NKCC1 is found in SCN neurons of the dorsal and ventral nuclei, regardless of time of day, and electrophysiologically recorded excitatory responses to GABA in the adult SCN disappear upon pharmacological inhibition of NKCC1 with bumetanide (Choi et al., 2008; Irwin and Allen, 2009; Belenky et al., 2010). Perhaps the most parsimonious view of this confusing body of evidence may be that GABA is primarily inhibitory during the day but that it has excitatory effects on a substantial number of SCN neurons at night (see Albers et al., 2017 for a review). Nevertheless, a significant number of GABA-excitable SCN neurons have been identified during the day (Liou and Albers, 1990; Wagner et al., 1997; Choi et al., 2008; Irwin and Allen, 2009; Alamilla et al., 2014).
Despite the important role of GABAAR signaling in phase shifting responses to both photic and nonphotic stimuli, the role of these CCCs in phase shifting the pacemaker is not known. Considering the prevalence of GABAAR signaling in SCN neurons, the role of GABAAR signaling in regulating the phase of the pacemaker, and the significant percentage of SCN neurons depolarized by GABA, it seems likely that excitatory responses to GABA play a functional role in entrainment within the intact SCN. The aim of these experiments was to test the hypothesis that NKCC1 activity within the SCN regulates both photic and nonphotic phase shifting in the fully intact circadian system.
Materials and Methods
Animals, Light Treatment, and Activity Monitoring
Adult male Syrian hamsters (approximately 9-10 weeks of age; 120-140 g) were purchased from Charles River Laboratories (Wilmington, MA) and individually housed in polycarbonate cages. All hamsters were held in a 14:10 light-dark (LD) cycle for 7 to 10 days (25 ± 1 °C) and allowed to acclimate to the animal facility and the LD cycle. Food (Lab Diet #5001; Nestlé Purina Pet Care, St. Louis, MO) and water were provided ad libitum. After the acclimation period, a single unilateral cannula was implanted aimed at the SCN in each hamster, and hamsters were allowed to recover from surgery (approximately days 8-15; LD 14:10). Next, hamsters were transferred to constant darkness (DD) for 10 to 14 days allowing for the establishment of free-running activity rhythms. All hamsters were held for a total of 15 to 20 days in LD 14:10 before release into constant conditions. The light stimulus used to induce phase delays and advances (CT 13.5 and CT 19, respectively) was a 150-lux 15-min light pulse (LP). Animal facility staff used a Kodak LED Safelight (660 nm) for routine animal husbandry. Each cage was equipped with a running wheel, and locomotor activity was monitored as wheel revolutions per 10 min and collected with VitalView Software (Mini Mitter, Bend, OR). All procedures conformed to National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee of Georgia State University.
Surgery and Drug Administration
Hamsters were deeply anesthetized (5% isoflurane induction; 2.5%-3.5% isoflurane for maintenance), placed into a stereotaxic device, and implanted with 4-mm, 26-gauge guide cannulas (PlasticsOne, Roanoke, VA) aimed at the SCN (0.8 mm anterior; 1.7 mm lateral to bregma, 10° angle toward midline). Microinjections were administered via a 33-gauge needle attached to polyethylene tubing (PE-20) and connected to a 1.0-µL Hamilton syringe. Microinjection volumes were 200 nL and were administered over 20 sec. The injection needle was left in place for 30 sec after injection to allow for diffusion of the solution. The depth of microinjection was 7.2 mm ventral to bregma. Injections and light pulses were given after approximately 10 to 14 days of exposure to DD. Bumetanide, dissolved in dimethyl sulfoxide (DMSO) as vehicle (Sigma Aldrich, St. Louis, MO), was used to inhibit NKCC1 chloride influx in order to decrease GABA-elicited excitatory responses in the SCN (Payne et al., 2003; Choi et al., 2008; Irwin and Allen, 2009). The solubility of bumetanide has been reported to range from 25 mM to more than 200 mM based on information from several different manufacturers and chemical societies. For the present study we were able to dissolve up to 1 M bumetanide in fresh (important due to the hygroscopicity of DMSO) 100% DMSO. Multiple doses, beginning with a low dose of 10 µM, were used to generate dose responses to bumetanide injected into the SCN region in vivo. Upon completion of each experiment, injection sites were verified with dye injection, histological preparation, and light microscopy. Unilateral injections of 200 nL spread slightly less than 1 mm from the injection site and, thus, diffuse to the contralateral SCN (Gillespie et al., 1999; Caldwell and Albers, 2003; Paul et al., 2005; Albers et al., 2017). Further, injections of GABA-active drugs 500 µm or more from the SCN do not induce phase shifts in activity rhythms (Hummer et al., 2015). The adult Syrian hamster SCN is around 0.6 mm × 0.3 mm × 0.6 mm (in the rostro caudal, mediolateral, and dorsoventral axes, respectively) (Lydic et al., 1982). Thus, injection sites further than 500 µm from the border of the SCN as well as sites showing SCN damage were excluded from further analysis.
Early and Late Subjective Night
To investigate the role of NKCC1 activity in light-induced phase delays at CT 13.5 and light-induced phase advances at CT 19, hamsters were microinjected with bumetanide (10 µM, 100 µM, or 1 mM) or vehicle into the SCN region immediately followed by a 15-min 150-lux LP or a sham LP (hamster removed from cage under safe light for drug injection) (Table 1). After treatment, hamsters were immediately returned to their home cages in DD, and wheel running was continually monitored for 10 to 14 more days.
Group sizes by time and treatment.

Bum = NKCC1 inhibitor bumetanide; LP = 150 lux, 15-min light pulse; Mus = GABAAR agonist muscimol (21.9 mM); Sham = sham light pulse.
Subjective Day
Because muscimol injection into the SCN during the subjective day (CT 6) induces phase advances in activity rhythms, we explored the role of NKCC1-mediated excitation of SCN neurons in these GABAAR-signaled phase advances in hamsters housed in DD (Smith et al., 1989; Huhman et al., 1995; Gribkoff et al., 1998; Mintz et al., 2002). This treatment mimics the phase advances induced by several nonphotic stimuli (Mrosovsky et al., 1992; Mrosovsky, 1996). At CT 6, hamsters were microinjected with bumetanide alone (10 µM, 100 µM, or 1 mM), vehicle, or the same concentrations of bumetanide in a cocktail with a phase-advancing dose of muscimol (21.9 mM) (Mintz et al., 2002) (Table 1). After injection, animals were returned to their home cages in DD, and wheel running was continually monitored for 10 to 14 more days.
Statistical Analysis
Phase shifts were quantified using the linear regression method (Daan and Pittendrigh, 1976). Regression lines were fit to activity onsets for 7 to 10 days, both for pretest baselines and postmanipulation onsets (ClockLab; Actimetrics, Wilmette, IL). Postinjection regression lines omitted 2 to 3 days of onsets following injection due to transient effects of light and handling on the free-running rhythm. Data are represented with mean (±SEM) phase shifts (hours) and were analyzed using 1-way ANOVA followed by a Tukey’s HSD post hoc test for significant ANOVAs. Significance was set at p ≤ 0.050.
Results
Effect of Inhibition of NKCC1 on Light-induced Phase Delays (CT 13.5)
Injection of bumetanide, an inhibitor of NKCC1, into the SCN region prior to a light pulse at CT 13.5 decreased the magnitude of light-induced phase delays (F3,23 = 3.135, p = 0.045). Tukey’s HSD post hoc testing revealed that 10 µM bumetanide decreased the magnitude of the phase delay when compared with vehicle (p = 0.027). The other two concentrations (100 µM and 1 mM) of bumetanide did not differ from vehicle or 10 µM bumetanide (Fig. 1). Injection of bumetanide alone (without light) at CT 13.5 did not produce phase shifts when compared with vehicle controls (F3,12 = 0.168, p = 0.916) (Fig. 2).
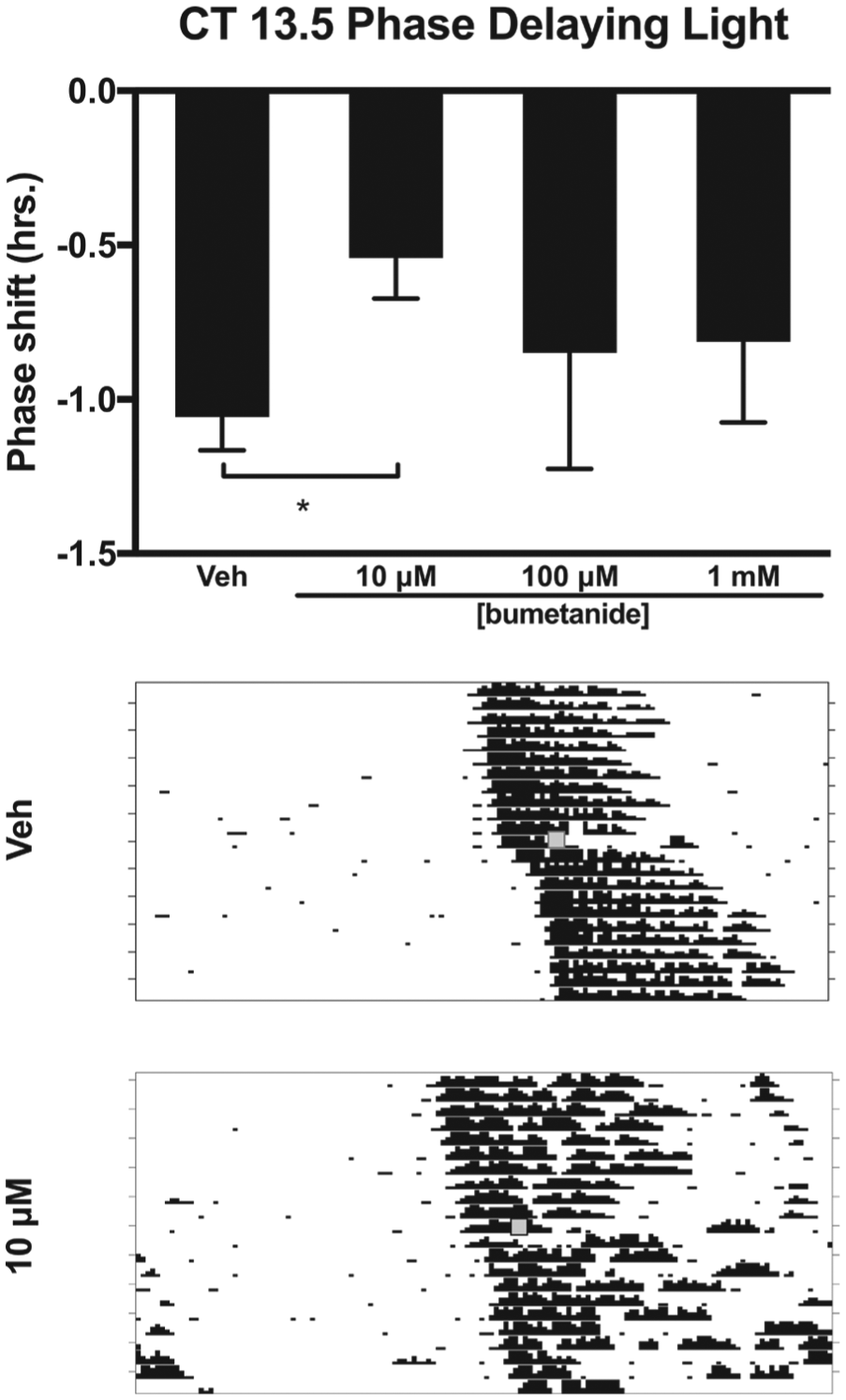
Effects of the NKCC1 inhibitor bumetanide injected into the SCN on light-induced phase delays of the circadian locomotor rhythm in the early subjective night (CT 13.5). Top: 10 µM bumetanide reduced phase delays in response to light at CT 13.5 compared with vehicle (*p ≤ 0.05). Middle and bottom: Representative wheel-running actograms for vehicle injection + light pulse and 10 µM bumetanide injection + light pulse, respectively. Gray boxes denote manipulation time point (i.e., drug treatment + light pulse).
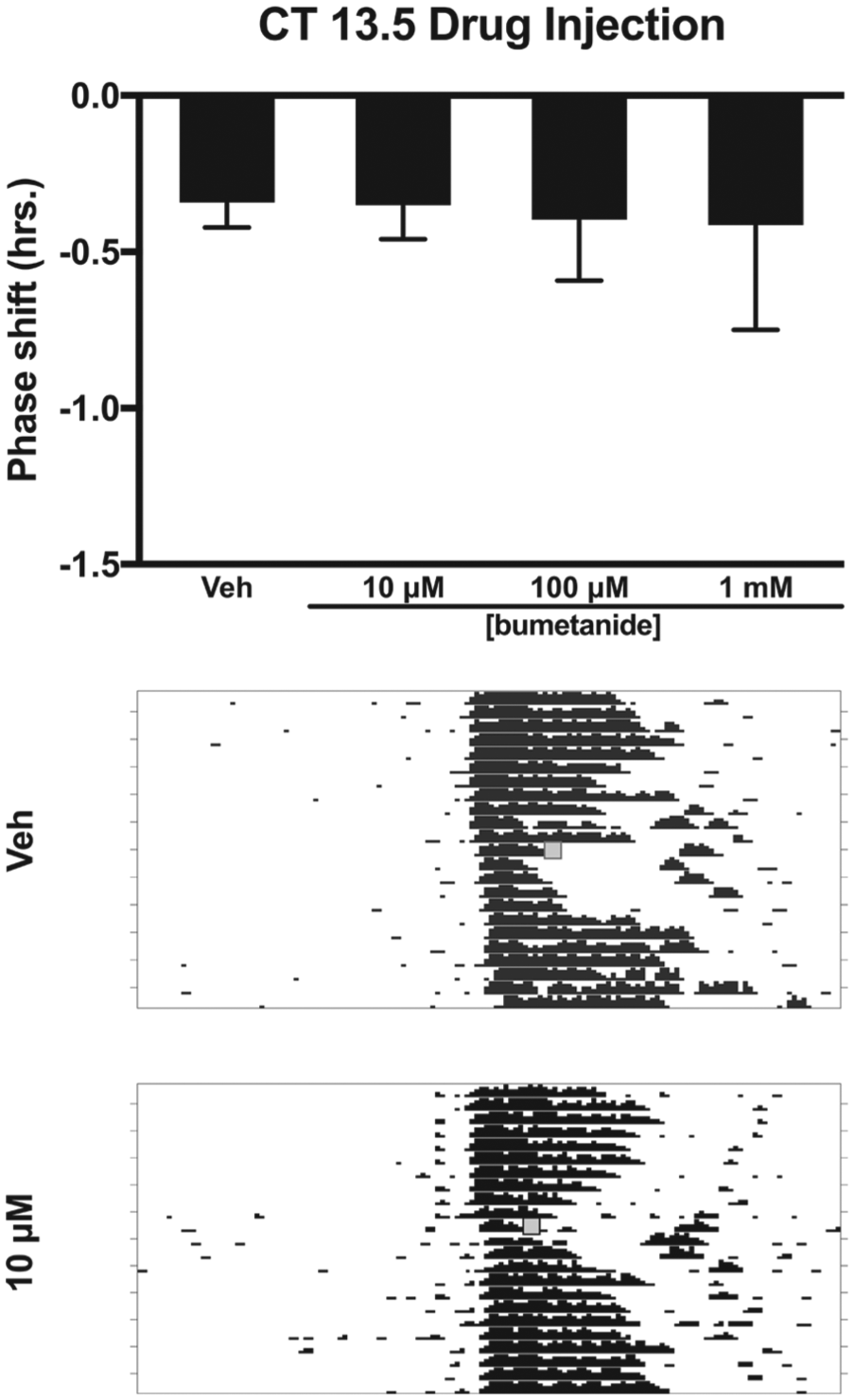
Effects of the NKCC1 inhibitor bumetanide injected into the SCN on the phase of the circadian locomotor rhythm in the early subjective night (CT 13.5). Top: Bumetanide did not produce phase shifts in the free-running locomotor rhythm at any concentration (p > 0.05). Middle and bottom: Representative wheel-running actograms for vehicle injection and 10 µM bumetanide injections, respectively. Gray boxes denote manipulation time point (i.e., drug treatment).
Effect of Inhibition of NKCC1 in the Late Subjective Night (CT 19)
During the late subjective night (CT 19), injection of bumetanide into the SCN region did not alter the magnitude of light-induced phase advances (F3,23 = 0.445, p = 0.723) (Fig. 3). Analysis of variance of bumetanide injected into the SCN region in the late subjective night (CT 19) in the absence of light produced a significant F statistic (F3,25 = 3.092, p = 0.045). However, Tukey’s HSD post hoc analysis found no effect of treatment on circadian phase. The 10 µM concentration of bumetanide trended toward a small advance of activity onset when compared with other treatments (p = 0.054-0.091) (Fig. 4).
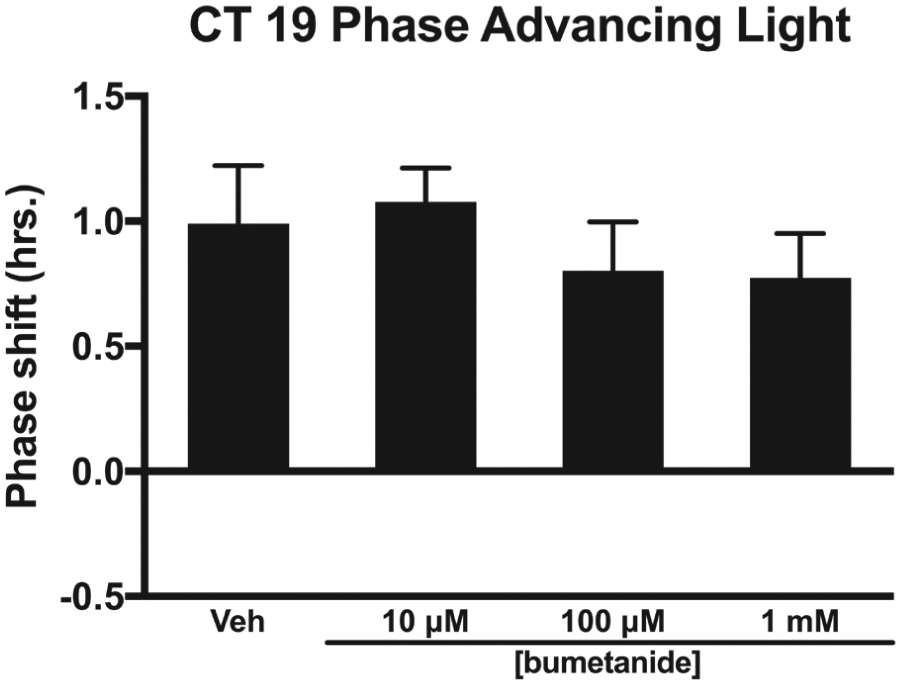
Effects of the NKCC1 inhibitor bumetanide injected into the SCN on light-induced phase advances of the circadian locomotor rhythm in the late subjective night (CT 19). Bumetanide did not alter the magnitude of light-induced phase advances at any concentration (p > 0.05).
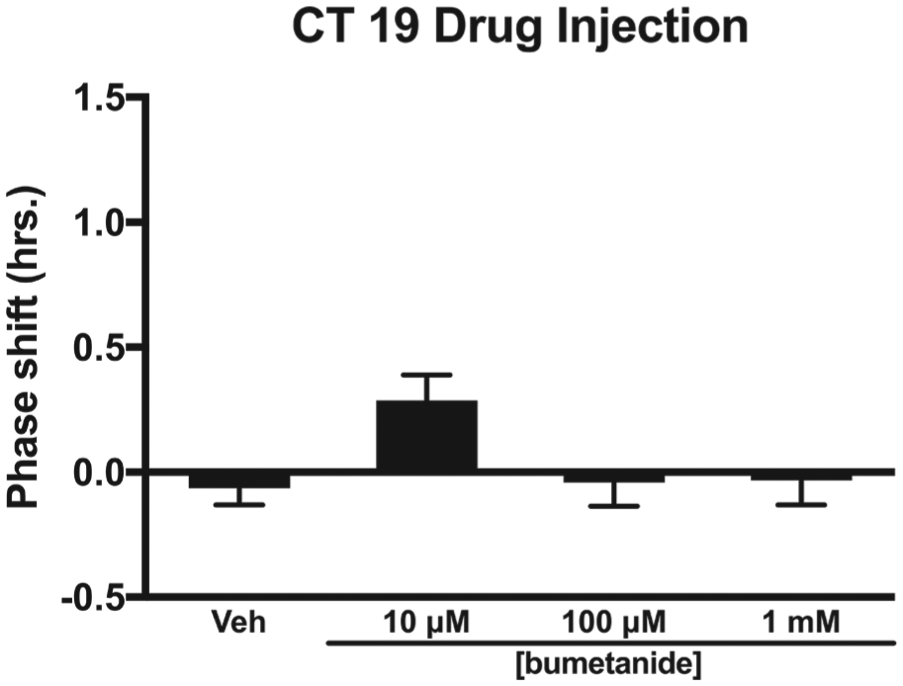
Effects of the NKCC1 inhibitor bumetanide injected into the SCN on the phase of the circadian locomotor rhythm in the late subjective night (CT 19). Bumetanide injection into the SCN did not phase shift the circadian locomotor rhythm in the late subjective night (CT 19) (p > 0.05).
Effect of Inhibition of NKCC1 during the Subjective Day (CT 6)
Next, we explored whether NKCC1 inhibition in the subjective day phase shifts the free-running activity rhythm. Injection of bumetanide alone into the SCN at CT 6 did not produce significant phase shifts at any concentration (F3,19 = 3.080, p = 0.052) (Fig. 5). We also examined whether NKCC1 activity modulates the phase advances produced by injection of the GABAAR agonist muscimol during the subjective day. We coadministered the same 3 concentrations of bumetanide used at CT 13.5 and CT 19 in a cocktail with a concentration of muscimol previously shown to produce phase advances during the subjective day (21.9 mM) (Mintz et al., 2002). Analysis of variance revealed a main effect of drug treatment (F4,27 = 4.152, p = 0.010). Tukey’s HSD post hoc testing revealed that muscimol (21.9 mM) administration led to a significant phase advance of the rhythm compared with controls (p = 0.010) (Fig. 6). A bumetanide concentration of 100 µM attenuated muscimol-induced phase advances (p = 0.024). Lower and higher concentrations of the NKCC1 inhibitor did not differ from muscimol administered alone (Fig. 6).
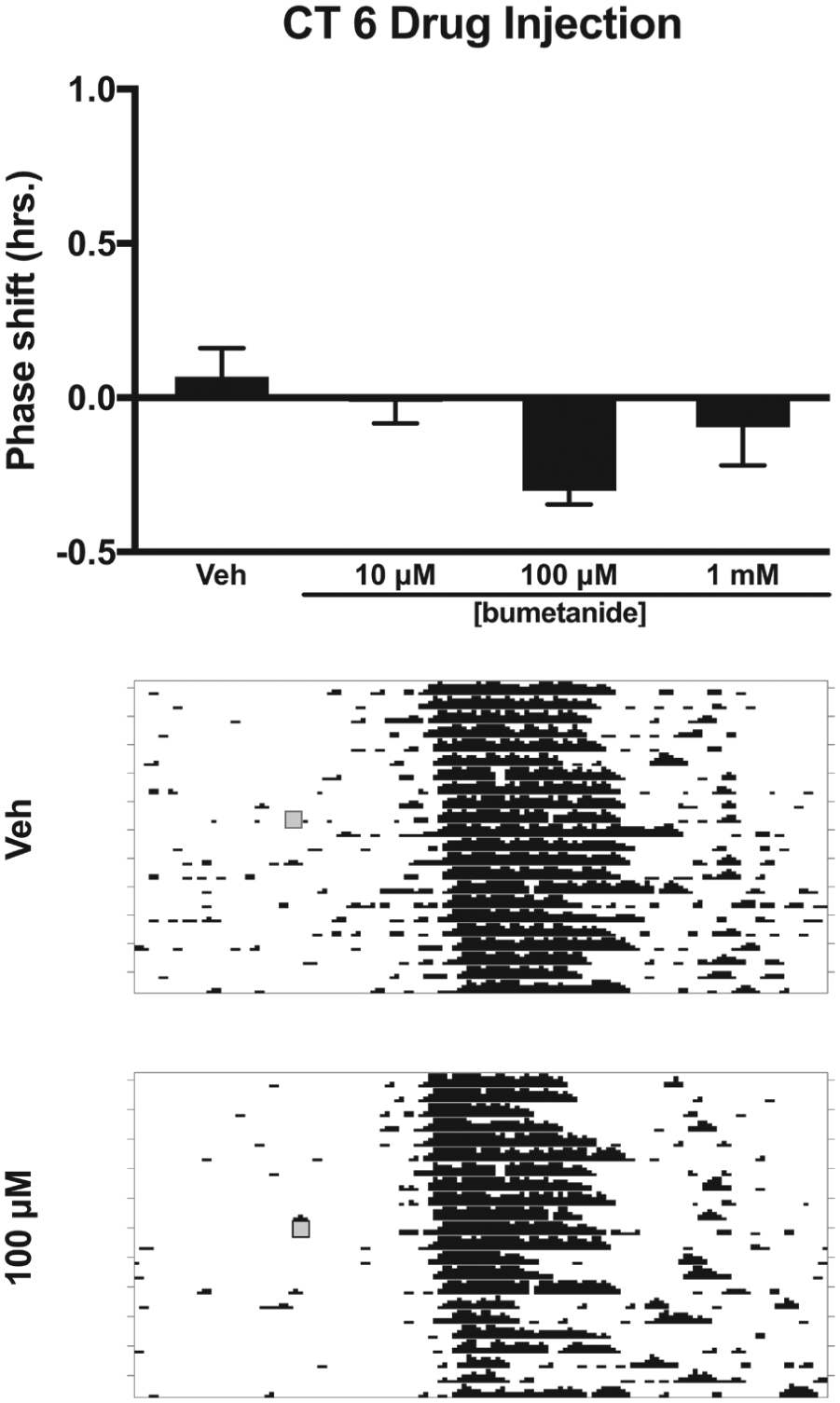
Effects of the NKCC1 inhibitor bumetanide injected into the SCN on the phase of the circadian locomotor rhythm during the subjective day (CT 6). Top: Bumetanide did not produce phase shifts in the free-running locomotor rhythm at any concentration (p > 0.05). Middle and bottom: Representative wheel-running actograms for vehicle injection and 100 µM bumetanide injections, respectively. Gray boxes denote manipulation time point (i.e., drug treatment).
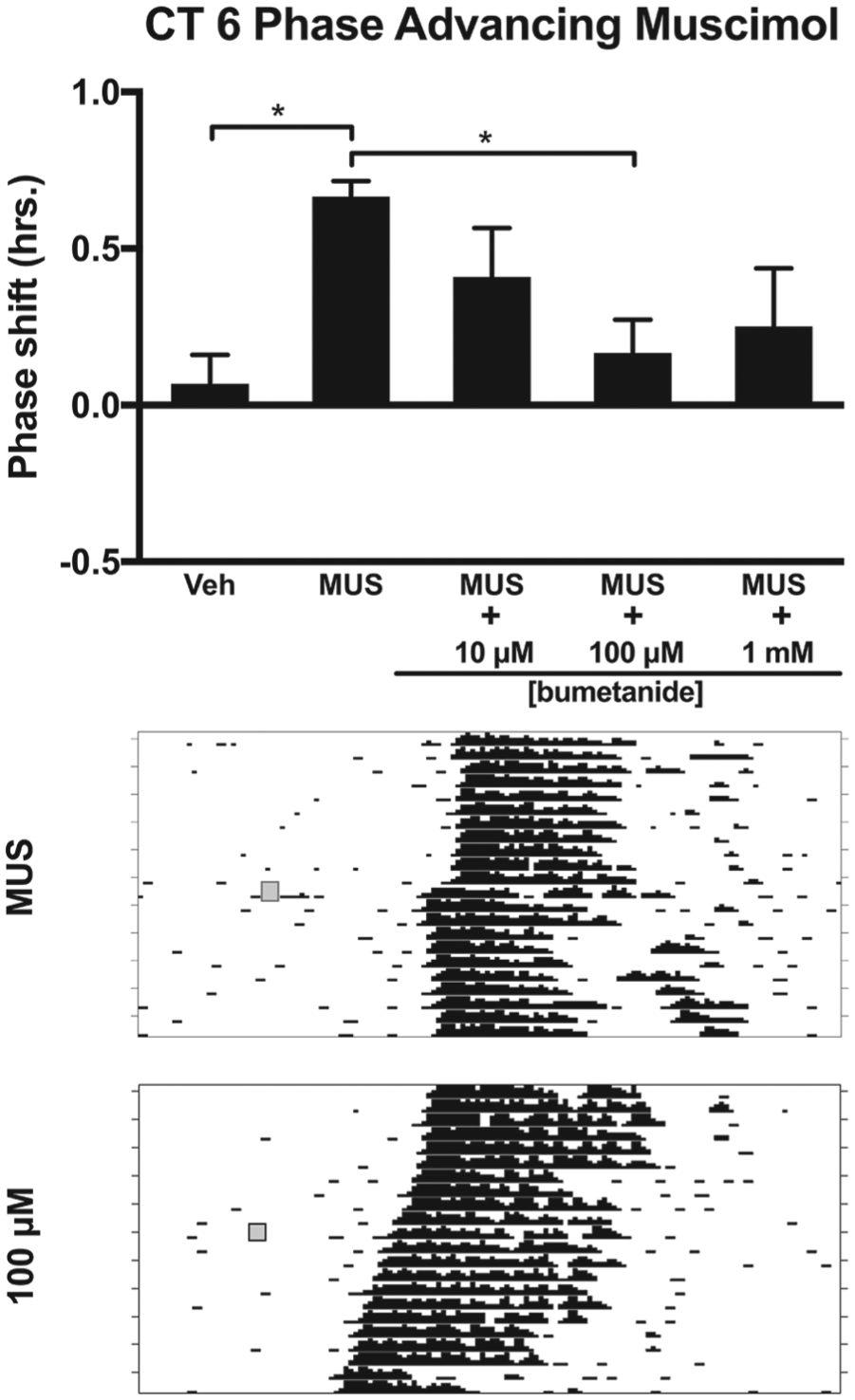
Effects of the NKCC1 inhibitor bumetanide injected into the SCN on muscimol-induced phase advances of the circadian locomotor rhythm during the subjective day (CT 6). Top: Muscimol administration into the SCN phase advanced the circadian locomotor rhythm compared with vehicle control (*p ≤ 0.05). Muscimol-induced phase advances were reduced by injection of 100 µM bumetanide (*p ≤ 0.05). Middle and bottom: Representative wheel-running actograms for 21.9 mM muscimol injection and 100 µM bumetanide + 21.9 mM muscimol injections, respectively. Gray boxes denote manipulation time point (i.e., drug treatment).
Discussion
These data indicate that injection of the NKCC1 inhibitor bumetanide within the SCN modulates the ability of the pacemaker to phase shift in response to both photic and nonphotic stimuli. In the early subjective night (i.e., CT 13.5), injection of bumetanide at a concentration (i.e., 10 µM) that inhibits SCN excitatory responses to GABA in vitro (Choi et al., 2008; Irwin and Allen, 2009; Alamilla et al., 2014; Farajnia et al., 2014; Kim et al., 2016) reduced the phase delays produced by light by approximately 50% (Fig. 1). Further support for the possibility that bumetanide reduces the ability of light to induce phase delays was the finding that bumetanide did not alter circadian phase in the absence of a light pulse at CT 13.5 (Fig. 2).
In the late subjective night (i.e., CT 19), injection of bumetanide into the SCN region did not reduce light-induced phase advances (Fig. 3). Furthermore, the injection of bumetanide into the SCN in the late subjective night in the absence of light did not lead to significant phase shifts of the rhythm at any concentration (Fig. 4). These differences suggest a different role for GABAAR-induced excitation in light-induced phase delays and phase advances or possibly no role for GABAAR excitatory responses in light-induced advances. Our laboratory has previously shown that the GABAAR agonist muscimol attenuates light-induced phase delays and advances in both night-active and day-active species (Gillespie et al., 1997; Novak and Albers, 2004). However, injection of the GABAAR antagonist bicuculline into the SCN region potentiates light-induced phase delays in the early night but not light-induced phase advances in the late night (Gillespie et al., 1997). In contrast, however, microinjection of bicuculline before phase-delaying or phase-advancing light pulses increases c-Fosimmunoreactivity within the SCN (Gillespie et al., 1999). Different GABAAR mechanisms may be involved in mediating light-induced phase delays and phase advances that may involve phase-dependent changes in the excitatory-inhibitory ratio of GABAAR containing SCN neurons and phase-dependent changes in GABAAR subunit composition (Choi et al., 2008; Irwin and Allen, 2009; Walton et al., 2017).
To determine whether the phase advances mediated by activation of GABAARs in the subjective day (i.e., CT 6) require NKCC1 activity, we examined the effects of bumetanide on muscimol-induced phase advances. As expected, injection of muscimol into the SCN region produced phase advances when compared with injections of vehicle (Fig. 6). Injection of bumetanide into the SCN in the subjective day did not induce phase shifts that differed from vehicle (Fig. 5). The injection of muscimol combined with bumetanide inhibited muscimol-induced phase advances when bumetanide was added at a concentration of 100 µM. No significant reductions in muscimol-induced phase advances were observed when bumetanide was coadministered at concentrations of 10 µM or 1 mM (Fig. 6).
The present experiments are the first to examine whether the NKCC1 inhibitor bumetanide influences the phase of the circadian pacemaker by its actions within the SCN in vivo. Prior work has focused on the effects of bumetanide on the cellular responses of SCN neurons monitored in vitro. These in vitro studies have found that micromolar concentrations of bumetanide (i.e., 10-100 µM) inhibit the excitatory effects of GABA within the SCN (Choi et al., 2008; Irwin and Allen, 2009; Alamilla et al., 2014; Farajnia et al., 2014; Kim et al., 2016). Bumetanide has a 500-fold greater affinity for NKCCs than KCCs; thus, low bumetanide concentrations (up to 100 µM) can be used in vitro to inhibit NKCCs without affecting KCCs, as observed in non-SCN brain tissue (Gamba, 2005). However, at higher concentrations, bumetanide begins to inhibit chloride efflux via actions on KCCs (Gamba, 2005; Delpire et al., 2009). These concentration-dependent, off-target actions of bumetanide on chloride efflux proteins (KCCs) may explain why we did not observe any circadian effects of 1 mM bumetanide at any time during the cycle. The effects of higher concentrations of bumetanide on SCN cellular activity in vitro, however, are not known as no dose-response work above 100 µM has been performed in the SCN, to our knowledge. Future work using microinjections of specific KCC2 inhibitors (e.g., VU024551) will be needed to address the functional role of KCC2 as well as other KCCs (1, 3, and 4) expressed in the SCN (Belenky et al., 2010; Klett and Allen, 2017).
Importantly, the ratio of GABA-induced excitation to inhibition in SCN neurons changes over the circadian cycle, as shown in multiple studies, although the exact temporal pattern of these changes is not clear, with reports of GABA-induced excitation occurring in the day, the night, or both (Wagner et al., 1997; Choi et al., 2008; Irwin and Allen, 2009; Alamilla et al., 2014; Farajnia et al., 2014). GABA excitation of SCN neurons may also differ in the ventral and dorsal subregions, although these findings are also not consistent. Dorsal SCN neurons are more likely to display excitatory postsynaptic potentials (EPSPs) upon GABA application, and NKCC1 protein may be higher in the dorsal SCN at night, although other authors have reported no differences (Choi et al., 2008; Belenky et al., 2010). Optic chiasm stimulation of the RHT or GABA application are more likely to increase the intracellular calcium concentration ([Ca2+i]) in the dorsal SCN (Irwin and Allen, 2009). Klett and Allen (2017) used chloride sensors to show that bumetanide causes small changes in intracellular chloride concentration ([Cl–i]). Surprisingly, bumetanide increases [Cl–i] in arginine-vasopressin–positive (AVP+) neurons and decreases [Cl–i] in vasoactive intestinal polypeptide–positive (VIP+) neurons. KCC antagonism increases [Cl–i] in both AVP+ and VIP+ SCN neurons, as expected, upon inhibition of the chloride efflux protein. In addition, bumetanide blocks the effects of a specific KCC2 antagonist on [Cl–i], suggesting that NKCC1 is regulating chloride influx in SCN neurons, as shown in other brain regions (Klett and Allen, 2017). Furthermore, we cannot rule out the possibility that our microinjection of NKCC1 inhibitor acted on non-AVP+ or non-VIP+ SCN neurons, as we applied the inhibitor to the entire SCN region and these cells comprise a small percentage of total SCN network neurons: VIP ≈ 10% and AVP ≈ 20% (Welsh et al., 2010). Inhibition of NKCC1 in non-AVP/VIP cells may be responsible for the modulation of phase shifts. Of course, the data of the present study cannot discriminate among these possibilities. However, it is important to note that most, if not all, of these neuropeptidergic cells colocalize with GABA in SCN neurons (Moore and Speh, 1993). It is also possible that our injections of bumetanide may have affected glycinergic signaling within the SCN, as glycine receptors also mediate a chloride current in SCN neurons and their activation may induce phase advances during the subjective day and phase delays in the early night (Ito et al., 1991; Mordel et al., 2011). However, our findings in the subjective day suggest that bumetanide inhibits phase advances directly signaled by the GABAAR, as we inhibited subjective day advances induced by the GABAAR agonist muscimol. In addition, to our knowledge, no in vivo work has explored the role of glycine in SCN mechanisms of entrainment to light.
Importantly, the differences observed in the present study suggest that the effects of bumetanide within the SCN are both phase and concentration dependent, consistent with observations made in vitro. Concentrations of bumetanide that dramatically inhibit the excitatory effects of GABA in vitro reduce the phase-delaying effects of light at CT 13.5. These data are consistent with a theoretical model in which endogenously released GABA is proposed to increase depolarization in response to RHT signaling in neurons with a higher ratio of NKCC1:KCC2 activity (i.e., excitatory to inhibitory) but to induce hyperpolarization in response to RHT signaling in neurons with a lower ratio of NKCC1:KCC2 activity (Irwin and Allen, 2009). The findings of the present study provide support for this model and significantly extend it by demonstrating that the excitatory effects of endogenously released GABA at CT 13.5 contribute to the ability of light to induce phase delays, thereby contributing to the most important function of the circadian system—its entrainment with the day-night cycle. Indeed, reduction of the excitatory effects of endogenously released GABA reduces the phase delay produced by light at CT 13.5 (Fig. 1).
Importantly, activation of the RHT produces GABA-induced excitatory responses in some SCN neurons and GABA-induced inhibitory responses in other SCN neurons (Irwin and Allen, 2009). Likewise, the present data indicate that GABA released in response to RHT activation enhances light-induced phase delays, while previous work has found that GABAA antagonists can enhance and GABAA agonists can inhibit light-induced phase delays at CT 13.5 (Gillespie et al., 1996). How can endogenously released GABA enhance light-induced phase delays and also inhibit light-induced phase delays? Perhaps these opposite effects of GABA are the result of GABAAR activation in different subregions or on subsets of neurons within the SCN with different NKCC1:KCC ratios. For example, the enhancement of light-induced phase delays could be mediated by the excitatory effects of GABA endogenously released in the dorsal SCN, whereas the inhibitory effects of GABA on light-induced phase delays could be mediated by activation of GABAARs in the retinoreceptive ventral SCN. The inhibitory effects of GABA on light-induced phase delays could be the result of the inhibition of RHT signaling to the ventral SCN and/or the inhibition of GABA release in neurons that project from the ventral to the dorsal SCN. In support of this idea are the findings that more NKCC1 activity is seen in the dorsal SCN and that the excitatory effects of GABA may be proportionally greater in the dorsal SCN than in the ventral SCN, particularly during the night (Choi et al., 2008; Irwin and Allen, 2009).
In addition to the acute effects of activating or inhibiting SCN GABAARs on the ability of light to induce phase delays, the sustained activation of GABAARs appears to be necessary for light to induce phase delays at CT 13.5 (Hummer et al., 2015). Because phase-delaying pulses of light produce a sustained activation of SCN neurons that lasts more than 4 h, and the SCN is a largely GABAergic network, it seems likely that there is a corresponding sustained release of GABA in the SCN (Hamada et al., 2001; Kuhlman et al., 2003; LeSauter et al., 2011; DeWoskin et al., 2015). Support for the hypothesis that the sustained activation of GABAARs in the SCN is necessary for the phase-delaying effects of light comes from the findings that injection of muscimol into the SCN beginning at CT 13.5 must occur over at least 4 h to mimic phase delays to light and that the injection of bicuculline must be given for 6 consecutive hours to block the phase-delaying effects of light (Hummer et al., 2015). Whether the ability of the sustained activation of GABAARs to produce phase delays is the result of excitatory, inhibitory, or bipolar responses to GABA is not known. One hypothesis that would be consistent with the existing data is that the sustained activation of GABAARs produces sustained excitatory responses (perhaps in the dorsal SCN) and the sustained excitatory responses are necessary to phase delay the pacemaker (Choi et al., 2008; DeWoskin et al., 2015; Hummer et al., 2015). If correct, this hypothesis might also explain why bumetanide inhibits light-induced phase delays in the present study because bumetanide would reduce the duration of the excitatory responses to GABA induced by light.
During the subjective day, bumetanide inhibited the ability of muscimol to induce phase advances, supporting the hypothesis that excitatory responses to GABA contribute to the phase-advancing effects of muscimol at CT 6 (Fig. 6). A functional role for the excitatory effects of GABA during the day is consistent with electrophysiological evidence reporting that GABA has primarily excitatory effects on SCN neurons during the day (Wagner et al., 1997; Alamilla et al., 2014). Even in several of the studies that found GABA’s excitatory effects to occur primarily during the night, a significant number of excitatory responses to GABA and muscimol were also observed during the day (Choi et al., 2008; Irwin and Allen, 2009). Because activation of GABAARs appears to be necessary for the phase shifting effects of NPY as well as other nonphotic phase shifting stimuli, the excitatory effects of GABA may be partially responsible for mediating these nonphotic shifts. Thus, these data provide further support for the hypothesis that GABA’s excitatory effects in the SCN play an important functional role in circadian timekeeping.
Footnotes
Acknowledgements
We thank Dr. Daniel Hummer and Alisa Norvelle for assistance with these experiments. We also thank Ancilla Titus-Scotland, Robert Bynes, and Christopher Barrow for animal husbandry. This work was supported by the National Institutes of Health grants R01NS078220 to H.E.A. and F32NS092545 to J.C.W.
Author Contributions
J.K.M., J.C.W., and H.E.A. designed and performed the research, analyzed the data, and wrote the manuscript.
Conflicts of Interest Statement
The author(s) have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.