
Abstract
The circadian rhythms found in almost all organisms are driven by molecular oscillators, including transcription/translation feedback loops (TTFLs). However, TTFL-independent oscillators can drive rhythms in both eukaryotes and prokaryotes. The fungus Neurospora crassa is a model organism for studying the molecular mechanism of the circadian clock. Although a circadian TTFL involving the proteins FRQ, WC-1, and WC-2 is well-characterized in N. crassa, rhythms can still be observed in the absence of this feedback loop. These rhythms are said to be driven by 1 or more FRQ-less oscillator(s) (FLOs). The prd-1 mutation lengthens the period in frq wild type and was previously shown to severely disrupt FRQ-less rhythms in frq null mutants under several different conditions; therefore, the prd-1 gene product is a candidate for a component of a FLO. We report here that prd-1 also disrupts free-running rhythms in wc-1 null mutants, confirming its effects on FRQ-less rhythms. We have now mapped and identified the prd-1 gene as NCU07839, a DEAD-box RNA helicase dbp-2. Complementation with the wild-type gene corrects the rhythm defects of the prd-1 mutant in the complete circadian system (when the FRQ-based TTFL is intact) and also the free-running FRQ-less rhythm on low choline. A PRD-1–GFP fusion protein localizes to the nucleus. The prd-1 mutant has a single base pair change in the first base of an intron that results in abnormally spliced transcripts. FRQ-less rhythms on low choline, or entrained to heat pulses, were only marginally affected in strains carrying deletions of 2 other RNA helicases (prd-6 and msp-8). We conclude that PRD-1 is a member of an RNA helicase family that may be specifically involved in regulating rhythmicity in N. crassa in both the complete circadian system and FLO(s).
Circadian (daily) rhythmicity, found in all kingdoms of life and at all levels of organization, is a fundamental property of cells. The molecular mechanisms of circadian (daily) rhythms have been studied for decades, and yet a complete description of any circadian system still eludes us. Transcription/translation feedback loops (TTFLs) are believed to form the core oscillators that drive rhythms in many organisms, although the identities of the key players differ between species, and evolutionary relationships between the various TTFLs are not obvious (Edgar et al., 2012). Rhythms can still be observed in the absence of these TTFLs, and the molecular mechanisms regulating these TTFL-independent oscillators remain poorly understood.
The filamentous fungus Neurospora crassa is one of the key model organisms in the search for circadian mechanisms. It was the second organism in which a TTFL was identified as an important molecular mechanism in the circadian system, and this TTFL continues to be intensively studied (for a recent review, see Cha et al., 2015). In this feedback loop, the FRQ (frequency) and WC-1 and WC-2 (white collar 1 and 2) proteins are key players. WC-1 and WC-2 form a transcription factor complex, WCC, which activates transcription of frq as well as a number of other genes. FRQ, along with its binding partner FRH (FRQ-interacting RNA helicase), inhibits its own transcription by promoting phosphorylation and inactivation of WCC. In the widely accepted TTFL model for circadian oscillators, the rhythmic activation/deactivation of the positive element (WCC in N. crassa) by oscillating levels of a negative element (FRQ/FRH in N. crassa) is assumed to drive the 24-h rhythms in observed outputs, such as the conidiation (spore-forming) rhythm of N. crassa.
It has been known for some time that the well-known TTFLs are inadequate to fully describe the circadian systems of many organisms (Lakin-Thomas, 2006b), and the importance of nontranscriptional oscillators is increasingly recognized (van Ooijen and Millar, 2012; Reddy and Rey, 2014). In a recent example, a central assumption of the N. crassa TTFL has been called into question. It has been assumed that the levels of the negative element FRQ must control the period of the feedback loop. However, it has now been demonstrated that even if this negative feedback element FRQ is not degraded, rhythms still persist (Larrondo et al., 2015). These observations suggest a circadian system more complex than the TTFL.
The most compelling evidence for a circadian system broader than the FRQ/WCC TTFL in N. crassa is the large and growing list of conditions and genetic backgrounds in which rhythms, both developmental (conidiation) and molecular, can be seen despite the absence of a functioning FRQ/WCC TTFL (reviewed in Lakin-Thomas et al., 2011). No clear and consistent picture has emerged of the genesis of FRQ-less rhythms, due to the variety of conditions that have been used to assay them. For example, frq null mutants can show conidiation rhythms in extra-long growth tubes (race tubes) (Loros and Feldman, 1986) or in genetic backgrounds such as vvd (Schneider et al., 2009) or sod-1 (Yoshida et al., 2008). Entrainment of frq null mutants to warm/cold cycles (Merrow et al., 1999; Roenneberg et al., 2005) or short heat pulses (Lakin-Thomas, 2006a) can elicit oscillator-like behavior in which the entrained phase is dependent on the T cycle. Molecular rhythms can be seen in frq null mutants, such as rhythms in nitrate reductase (Christensen et al., 2004) or gene transcription of ccg-16 and levels of WC-1 protein (de Paula et al., 2006). The most recent example is in the cog-1 mutant background, in which conidiation rhythms are seen in LL and in frq and WCC null mutants, which appear arrhythmic in a cog-1 wild-type background under similar conditions (Nsa et al., 2015).
The rhythms listed above must be driven by 1 or more oscillator(s) that can function without a complete FRQ/WCC TTFL, and these putative oscillators have been named FRQ-less oscillators or FLOs (Iwasaki and Dunlap, 2000). Because these various FRQ-less rhythms have been assayed by different laboratories under different conditions in different genetic backgrounds, it is not yet possible to determine whether multiple FLOs drive these rhythms or whether a single FLO could account for all or most of the observed rhythms. No molecular mechanisms have been described for any putative FLOs.
In our lab, we use 2 different assays to reveal FRQ-less rhythms in N. crassa: entrainment to cyclic heat pulses (Lakin-Thomas, 2006a) and free-running rhythms in constant temperature in choline-depleted cultures (Lakin-Thomas, 1996, 1998; Lakin-Thomas and Brody, 2000). (Note that the heat pulse entrainment assay does not depend on the chol-1 mutant.) In both assays, strains with null mutations in frq or wc-1 can be shown to display rhythmic behavior indicative of a functional oscillator. We have found that 3 different mutations (prd-1, prd-2, and UV90) affect both of these FRQ-less rhythms (Li and Lakin-Thomas, 2010; Li et al., 2011). We interpret this as evidence that there may be one FLO that drives both of these FRQ-less rhythms.
These 3 mutations (prd-1, prd-2, and UV90) also affect rhythmicity when the FRQ/WCC TTFL is functional: prd-1 and prd-2 affect the period of the conidiation rhythm (Feldman, 1982), and UV90 reduces the amplitude of both the conidiation rhythm and the FRQ protein rhythm (Li et al., 2011). These mutations therefore have effects on the complete circadian system, not just on FLO(s). This evidence demonstrates interactions between the FRQ/WCC TTFL and FLO(s). Other lines of evidence also demonstrate interactions: the cel mutant strain and the choline-depleted chol-1 strain express long-period conidiation rhythms that continue in frq-null strains, and in both assays, it was found that other alleles at the frq locus affect the long periods (Lakin-Thomas and Brody, 1985; Lakin-Thomas, 1998).
The prd-1 mutation (originally named frq-5) was identified based on its effect on the period of the conidiation rhythm (Feldman and Atkinson, 1978). It also decreases growth rate (Feldman and Atkinson, 1978). The prd-1 mutation affects 4 different FRQ-less rhythms. First, it blocks the period-lengthening effects of exogenous fatty acids on the conidiation rhythm in fatty acid–requiring cel strains (Lakin-Thomas and Brody, 1985), and this long-period cel rhythm was later shown to be driven by a FLO (Lakin-Thomas and Brody, 2000). Second, when farnesol is added to the growth medium, a FRQ-less conidiation rhythm is seen in frq null strains, and the prd-1 mutation lengthens the period of this FRQ-less rhythm (Lombardi et al., 2007). Third, prd-1 severely disrupts the heat-entrained FRQ-less rhythm (Li and Lakin-Thomas, 2010). Fourth, prd-1 abolishes the free-running FRQ-less rhythm in choline-depleted chol-1 (Li and Lakin-Thomas, 2010).
The discovery that prd-1 affects 4 FRQ-less rhythms may indicate that a single FLO drives these 4 different observed rhythms (Lakin-Thomas et al., 2011) or that there are 4 different oscillators driving these 4 rhythms, and prd-1 plays an independent role in all of them. In the absence of evidence that these rhythms are independent phenomena, and in the spirit of Occam’s razor, we propose the simplest hypothesis that can explain the results, which is that a single FLO may drive all 4 of these rhythms. In this article, we will use the term FLO to indicate the oscillator that we propose drives these 4 FRQ-less rhythms under the assay conditions in our laboratory. We do not have evidence as to whether or not any other FRQ-less rhythms may be driven by the same FLO. Based on the evidence cited above for interactions between FRQ/WCC and FLO(s), our model for the complete circadian system (Li and Lakin-Thomas, 2010; Lakin-Thomas et al., 2011) includes the FRQ/WCC TTFL and at least 1 FLO mutually interacting to generate the output rhythms.
Understanding the mechanism of the FLO (or FLOs) is essential to understanding the circadian system as a whole. To that end, our goal in this work was to map and identify the gene responsible for the prd-1 phenotype. We found that the prd-1 gene is a member of the family of DEAD-box RNA helicases that may have a specific role in the circadian system.
Materials and Methods
Strains Used
The prd-1; rasbd strain (Fungal Genetics Stock Center [FGSC] 4902) was obtained from the Fungal Genetics Stock Center (Kansas State University, Manhattan, KS) and crossed into our laboratory strains to construct csp-1; prd-1; rasbd (Li and Lakin-Thomas, 2010). The Mauriceville wild type was FGSC 2225. The prd-2 strain used for sequencing was rasbd; prd-2 (FGSC 4903). Strains carrying gene deletions produced by the Neurospora Functional Genomics Project (Colot et al., 2006) were obtained from FGSC. The strains used for assaying rhythmic phenotypes of prd-6 and msp-8 were constructed by crossing the deletion strains FGSC 11230 (prd-6KO) and FGSC 12359 (msp-8KO) to a csp-1; chol-1 rasbd; frq9 strain constructed in our laboratory. Other strains were produced in our laboratory by standard crossing techniques as previously described (Lakin-Thomas and Brody, 2000). Genotypes of multiple mutant strains were identified by various criteria: rasbd by slower growth and banding phenotype, csp-1 by failure of conidial separation, chol-1 by slow growth without choline supplementation, prd-1 by slow growth and long period, frqnull and wc-1null by arrhythmic conidiation on high choline and presence of banding rhythms on zero choline in chol-1 strains, and gene deletions by hygromycin resistance (produced by the cassette used for gene replacement [Colot et al., 2006]) or by polymerase chain reaction (PCR) with gene-specific primers to confirm gene replacement. When necessary, multiple mutant strains were backcrossed to confirm segregation of the mutations.
The chol-1 mutation is carried in most of our strains so that we can assay the FRQ-less rhythm in choline-depleted cultures. When chol-1 is supplemented with choline, the chol-1 defect is repaired and the cultures are indistinguishable from chol+ strains in our assay system. This allows us to assay other FRQ-less rhythms that do not depend on choline depletion, such as heat pulse entrainment behavior, in the same genotypes.
Measurement of Growth, Period, and Heat Entrainment
Cultures were inoculated onto solid agar medium in 30-cm glass tubes (“race tubes”). Growth medium (MA) was 1× Vogel’s salts with 0.5% maltose, 0.01% arginine, and 2% agar. Choline was added at 100 µM when indicated. The race tubes were placed in light at 30 °C for 1 day and then transferred to constant darkness at 22 °C. The growth front was marked every day under red safelight, and the growth rate and period were calculated as previously described (Lakin-Thomas, 1998).
Entrainment to heat pulses in Supplemental Figure S5 was carried out as previously described (Li and Lakin-Thomas, 2010). Cultures were grown at 22 °C in constant darkness and given 2-h pulses of 32 °C every T hours, depending on the imposed T cycle. Conidiation density traces for 5 or 6 replicate tubes were averaged, using the last 2 or 4 cycles of each tube, and averaged traces were normalized to set the maximum density equal to 1.0.
Mapping of the prd-1 Mutation
Mapping cross
Using standard crossing methods, a cross was made between a laboratory-constructed prd-1 strain in the Oak Ridge (OR) background (csp-1; prd-1; rasbd) and the Mauriceville (MV) wild type. Progeny from this cross were phenotyped for csp-1 (conidial separation-1) by tapping the tubes to look for failure of spore separation and were inoculated onto race tubes to assay growth rate and conidial banding period to determine rasbd and prd-1 phenotypes.
CAPS markers
Initial mapping efforts used cleaved amplified polymorphic sequence (CAPS) markers described in Jin et al. (2007). Additional markers (K2T2 and P21Q21) were developed using the methods of Jin et al. (2007) as follows (see Suppl. Table S1). Sequence in the region of interest was manually scanned for restriction sites, and PCR primers were designed to amplify a region of about 400 bp with the restriction site near the center. Genomic DNA from OR and MV strains was amplified with the primers, and PCR products were cut with the restriction enzyme and run on agarose gels. Differences in the band sizes between OR and MV indicated the presence of a single-nucleotide polymorphism (SNP) somewhere in the restriction site. Exact SNP locations could not be determined with this method but were later identified once the Mauriceville sequence became available (Pomraning et al., 2011) (see Suppl. Table S1). The K2T2 SNP could not be identified due to the poor quality of the MV sequence in that region.
Subsequently, mapping used markers developed from lists of SNPs identified by Pomraning et al. (2011) and posted on the website of the Fungal Genetics Stock Center (http://www.fgsc.net/Neurospora/SNPs/SNP_map.htm). Primers were designed to amplify a region of about 400 bp with the SNP and restriction site near the center (see Suppl. Table S1). The presence of a restriction site in the PCR product was visualized as 2 additional bands on the gel; the absence of the restriction site due to the reported DNA polymorphism produced a single band (see Suppl. Table S1).
Cloning the prd-1 Gene and N. crassa Transformation
The genomic sequence of NCU07839 plus 189 bp upstream and 104 bp downstream was amplified from the DNA of a prd-1 wild-type strain by PCR using the following primers: forward, AATTGAATTCATCCGCATCTCACATCCACTAGC; reverse, CTCATCGACGTGAAACCTACAGC. The PCR product was cloned into the pSTblue-1 plasmid (EMD Millipore, Etobicoke, Ontario, Canada) by blunt-end ligation according to the manufacturer’s instructions. The insert was removed from this plasmid using EcoR1 and subcloned into the his-3 targeting plasmid pBM61 (Margolin et al., 1997). The coding sequence of NCU07839 (without upstream and downstream sequences) was amplified by PCR from the pBM61 plasmid carrying the NCU07839 gene using the following primers: forward (SpeI), GGACTAGTATGTCCGGATCTTACGGCGGC; reverse (PacI), CCTTAATTAACCACCGGCGGTTGTTAAGCGGA. The PCR product and plasmid pCCG::C-GLY::3XFLAG (Honda and Selker, 2009) were cut with SpeI and PacI and ligated to insert the coding sequence of NCU07839 between the highly expressed ccg-1 promoter and the 10× GLY linker site. The plasmid was introduced into the his-3 locus of a csp-1 his-3; prd-1; chol-1 rasbd strain by electroporation of conidiospores.
Purification of Homokaryons
Because conidiospores are usually multinucleate, transformants are likely to be heterokaryotic and were therefore purified to homokaryons by production of uninucleate microconidia using a method adapted from Pandit and Maheshwari (1993) and Ebbole and Sachs (1990). Insertion of transgenes at the his-3 locus and purification of transformants to homokaryons were confirmed by PCR using the following primers specific for the his-3 locus: forward, CTCATGTTCAACCCTTTGGATGG; reverse, GTCATGCAGTTGGCTAAGGTTGAG.
For purification of homokaryons from the heterokaryotic knockout of NCU07839, microconidia were plated on sorbose media containing hygromycin. Surviving colonies were grown on race tubes without hygromycin selection to allow proliferation of any hygromycin-sensitive nuclei. After growth, their genotypes were assayed by PCR using primers specific for the NCU07839 gene sequence: forward, GGTTCTTGTCTCTAGACGTC; reverse, CCTGTAGATGAGACTTTCCG.
Construction of GFP Fusion and Fluorescence Microscopy
The coding sequence of NCU07839 was amplified by PCR from the same pBM61 plasmid described above for construction of the FLAG-tagged gene using the SpeI and PacI primers. The PCR product and plasmid pCCG::C-GLY::GFP (Honda and Selker, 2009) were cut with SpeI and PacI and ligated to insert the coding sequence of NCU07839 between the highly expressed ccg-1 promoter and the 10× GLY linker site. The plasmid was transformed into a csp-1 his-3; prd-1; chol-1 rasbd strain, and transformants were purified by microconidia isolation as described above and verified by PCR.
A strain expressing the histone-H1-RFP fusion protein was used as a positive control for nuclear localization. To construct this strain, a plasmid expressing H1-RFP (Pccg-1–hH1+–tdimerRedN) was introduced into the his-3 locus of a csp-1 his-3; chol-1 rasbd; inl strain. This plasmid was constructed by amplifying the sequence corresponding to RFP from pMF334 (Pccg-1–tdimerRedN) (Freitag and Selker, 2005) and subcloning it into the PacI and EcoRI site of plasmid pMF280 (Pccg-1–hH1+–sgfp+) (Freitag et al., 2004b) to replace the sequence for GFP.
For differential interference contrast (DIC) and fluorescence microscopy, cultures of both transformant strains were grown overnight at 30 °C in constant light in 10-cm Petri dishes on Vogel’s N medium containing 2% glucose and 1.5% agar and supplemented with 100 µM choline and 200 µg/mL inositol. Square agar blocks (~1 cm) with growing hyphal tips and mycelia were cut out using coverslips, mounted onto glass slides, covered with ~100 µL liquid media (composition similar to agar medium), and then covered with a coverslip. Slides were visualized using an oil immersion lens (63×) in a Leica (Leica Microsystems, Richmond Hill, Ontario, Canada) DM5000 Fluorescence Microscope. Images were captured using Adobe Photoshop CS5 (Adobe, San Jose, CA) and analyzed with ImageJ (National Institutes of Health, Bethesda, MD) software.
For confocal microscopy, the PRD-1–GFP transformant was grown on microscope slides on MA medium with 3% agar and 100 µM choline for 24 h in constant light at 22 °C. Liquid medium (MA without agar) was added to the growing front under a coverslip, and images were captured from actively growing hyphae using a 60× oil immersion objective in a Bio-Rad (Hercules, CA) MRC 600 confocal laser scanning microscope. Image contrast was adjusted in Photoshop.
DNA Isolation and Sequencing
Cultures were grown at 30 °C in constant light for 2 or 3 days in standing liquid cultures in 24-well plates with 1 mL liquid medium per well (1× Vogel’s minimal medium with 2% glucose and 215 µM choline). Mycelial pads were harvested, washed in water, partially dried under vacuum, and frozen in liquid N2. Samples were powdered under liquid N2 and DNA was extracted as described in Jin et al. (2007).
Sequences from the Broad N. crassa database (FungiDB, http://fungidb.org/fungidb; Stajich et al., 2012) were used to design PCR primers to amplify the regions of interest from genomic DNA. Sequencing primers were designed every 500 to 800 nt. The gel-purified PCR products and primers were sent to the York Biology Molecular Core Facility for sequencing, and results were aligned with the Broad database genome sequence using ApE software (A plasmid Editor by M. Wayne Davis, http://biologylabs.utah.edu/jorgensen/wayned/ape/).
RNA Isolation and Complementary DNA Analysis
A prd-1 wild-type strain (csp-1; chol-1 rasbd) and a prd-1 mutant (csp-1; prd-1; chol-1 rasbd) were grown at either 22 °C or 30 °C in constant light for 3 days in standing liquid cultures as described above for DNA extraction. Samples were powdered under liquid N2 and RNA was extracted with Tri-Reagent (Molecular Research Center (Cincinnati, Ohio, USA)) according to the manufacturer’s directions. For complementary DNA (cDNA) preparation, the RNA was first treated with DNAse and reextracted with Tri-Reagent. cDNA was then synthesized using the SuperScript cDNA Synthesis Kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s directions. PCR was carried out on the cDNA to amplify the region containing the third intron, using the following primers located in the third and fourth exons: forward, CCCAACAACTCCGAGGATTAC; reverse, ACCTCCACCACCACTGTAGC (see Suppl. Figure S1A). The PCR products were cloned into the pJET 1.2 blunt-end cloning vector (CloneJET PCR cloning kit; Thermo Fisher (Waltham, MA, USA)) and sequenced with pJET forward and pJET reverse primers. Five clones from the wild type and 15 clones from the mutant were sent to BioBasic (Markham, Ontario, Canada) for sequencing. Sequences were aligned using ApE software (M. Wayne Davis, University of Utah, Salt Lake City, Utah, USA).
Results
prd-1 Mutation Disrupts FLO Rhythms in wc-1 Mutants
We have previously reported (Li and Lakin-Thomas, 2010) that the prd-1 mutation disrupts the conidiation rhythm in frqnull strains. To determine whether this mutation also abolishes FRQ-less rhythms in other strains lacking a functional FRQ/WCC TTFL, we constructed double mutants carrying both the prd-1 mutation and wc-1 null mutations, either wc-1 allele ER53 or wc-1ko. As shown in Figure 1, the free-running rhythm seen in choline-depleted cultures, in both wc-1 wild-type and wc-1null strains, is severely disrupted by the introduction of the prd-1 mutation. These results are very similar to those seen in the frqnull strains (Li and Lakin-Thomas, 2010). This confirms that when the FRQ/WCC TTFL is disabled, by ablating either FRQ or WC-1 function, the FRQ-less oscillator that drives these free-running rhythms remains functional, but it is dependent on the PRD-1 gene product to sustain it.

Effects of the prd-1 mutation on the rhythmic phenotype of wc-1 null mutants. Strains of the indicated genotypes (left) were grown on media with (+) or without (–) choline (100 µM). Two replicate race tubes are shown for each condition. All strains carry the rasbd, csp-1, and chol-1 mutations. White bars indicate the mean distance for 24 h of growth. The 2 alleles for wc-1 are ER53 and wc-1KO; similar results were obtained with multiple isolates of both wc-1 alleles (data not shown).
Mapping of prd-1
The prd-1 mutation, originally named frq-5, was obtained by mutagenesis of the rasbd strain with N-methyl-N′-nitro-N-nitrosoguanidine in the Feldman laboratory and was mapped to a position near the centromere on LG III (linkage group III, identified with chromosome III) (Feldman and Atkinson, 1978). These authors reported recombination frequencies of about 5% with acr-2 and about 20% with pro-1 (see Figure 2 for all markers used in mapping). For our mapping work, we took advantage of the well-established method of mapping the gene of interest relative to DNA polymorphisms identified between 2 strains of N. crassa, OR and MV (Metzenberg et al., 1984). The prd-1 mutation was originally generated in the OR genetic background (Feldman and Atkinson, 1978), and we have used the same genetic background in all our laboratory crosses. For mapping, we crossed a csp-1; prd-1; rasbd strain to the MV wild type. Among the csp-1 progeny, 4 clear classes could be discerned by growth rate and banding phenotypes: (1) csp-1, (2) csp-1; prd-1, (3) csp-1; rasbd, and (4) csp-1; prd-1; rasbd. Only the last 2 classes were used in mapping and will be referred to as prd-1 wild type and prd-1 progeny.
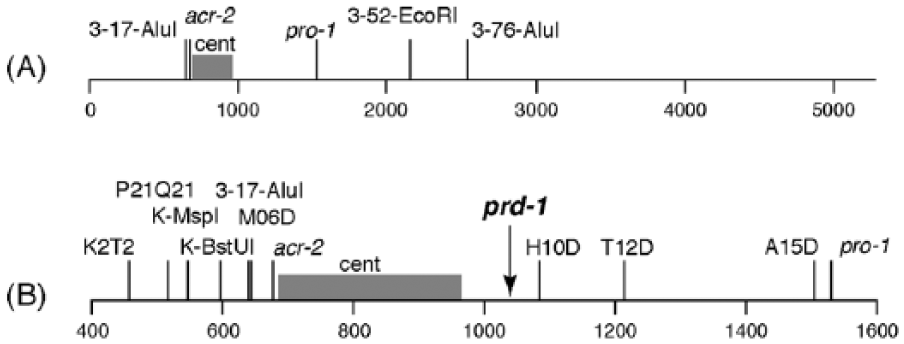
Mapping the prd-1 mutation on the right arm of linkage group III. (A) Markers on LG (linkage group) III. (B) Fine mapping using additional cleaved amplified polymorphic sequence (CAPS) markers in the expanded region of LG III. The numbers at the bottom indicate distance in kb on LG III. Conventional genetic markers are indicated in lowercase italics (e.g., pro-1), and CAPS markers are indicated in Roman font (e.g., 3-52-EcoRI). Position of the centromere is indicated by “cent.”
In our initial mapping, we used the CAPS markers and bulked segregant methods of Jin et al. (2007). In this method, the CAPS contain an SNP that affects a restriction enzyme site such that the site will be cleaved by the enzyme in one genetic background but not in the other. A short PCR product is designed with the SNP in the middle, and after restriction enzyme digestion, the PCR product is run on a gel to reveal either 2 cut fragments or 1 uncut fragment. For the bulked segregant method, approximately 40 progeny of each genotype (prd-1 wild type or prd-1) were chosen, DNA was prepared from individual progeny, and 2 DNA pools were made (prd-1 wild type or prd-1). These pooled DNA samples were used as templates for the PCR amplification of the region with the SNP. After enzyme digestion, the relative intensities of the resulting DNA bands give a semiquantitative estimate of the relative frequencies of the 2 genotypes in the pooled DNA (Jin et al., 2007). We chose LG III markers 3-17-AluI, 3-52-EcoRI, and 3-76-AluI from Jin et al. (2007) (Figure 2A). Results of bulk segregant analysis showed about 10% to 30% recombination with 3-52-EcoRI and 3-76-AluI and no recombination with 3-17-AluI.
Both 3-52-EcoRI and 3-76-AluI are to the right of 3-17-AluI, so we developed additional markers to the left. (All CAPS markers reported here are listed in Suppl. Table S1.) Markers K2T2 and P21Q21 were used, along with 3-17-AluI, to individually genotype 84 of the original progeny plus an additional 48, for a total of 132. K2T2 and P21Q21 gave recombination frequencies of 12.1% and 10.6%, putting prd-1 to the right of these markers (see Suppl. Table S2). The 3-17-AluI CAPS marker showed no recombination, putting prd-1 less than 2 map units from 3-17-AluI.
Another round of marker development produced 4 more CAPS markers—M06D, H10D, T12D, and A15D—to the right of 3-17-AluI. There was no recombination with M06D in 50 progeny and increasing recombination rates with the other markers using 132 progeny, putting prd-1 somewhere between markers P21Q21 and H10D (Figure 2B and Suppl. Table S2). To further refine the interval, 2 additional markers were developed, K-MspI and K-BstUI, and these were used to genotype a selection of progeny previously shown to have a recombination event somewhere in this interval. Recombinants were found with K-MspI but not K-BstUI, locating prd-1 to the right of K-MspI and to the left of H10D. Supplemental Table S3 summarizes the data obtained for all recombinant progeny.
Identification of the prd-1 Gene
The interval between K-MspI, at 545271, and H10D, at 1083480, encompasses 538,209 bp and approximately 66 genes. The centromere, devoid of functional genes, is located at approximately 682,000 to 966,000 (Smith et al., 2011), leaving approximately 250,000 bp in which prd-1 was likely to be located. We initially concentrated our search to the left of the centromere, near the marker 3-17-AluI at about 639,000, which showed no recombination with prd-1 in 132 progeny. We sequenced a number of likely open reading frames (ORFs) in the region 601,000 to 674,000, which were chosen based on annotations indicating critical functions, without identifying any sequence differences between prd-1 and prd-1 wild type (data not shown).
We then altered our approach to look at the phenotypes of strains carrying deletions of genes in the region, reasoning that the slow-growth phenotype of prd-1 could be used to identify candidates from the deletion collection, assuming the deletion is viable. (Note that the deletions did not carry the rasbd mutation and therefore they did not display a banding phenotype that would easily allow period calculations.) Deletion strains produced by the Neurospora Functional Genomics Project (Colot et al., 2006) were obtained from the FGSC and were screened for growth rate on race tubes. Among 22 deletions on the left of the centromere, only 1 (NCU05703) had a slow growth rate (about 65% of the wild-type growth rate), which is a characteristic of the prd-1 mutation (see Suppl.Table S4). An additional 19 deletions to the right of the centromere were screened, and one, NCU07817, grew at 56% of the wild-type rate (see Suppl. Table S4). These 2 ORFs plus about 200 to 400 bp upstream and downstream were sequenced, but no sequence differences were found between prd-1 and prd-1 wild type.
Not all genes in this interval were available as deletions, and a selection of likely candidates was chosen for sequencing. After sequencing of 3 additional genes, a mutation was found in NCU07839 in the prd-1; rasbd strain (FGSC 4902) (Suppl. Figure S1A,B and Suppl. Table S4). An additional lab strain, csp-1; prd-1; rasbd, also carried the mutation, but it was not found in csp-1; rasbd. Three prd-1 progeny from the mapping cross to Mauriceville and 2 additional prd-1 lab strains also carried the mutation, and 3 prd-1 wild-type progeny plus 3 prd-1 wild-type lab strains did not, confirming that the mutation is specific to the prd-1 phenotype.
Complementation of the prd-1 Mutation.
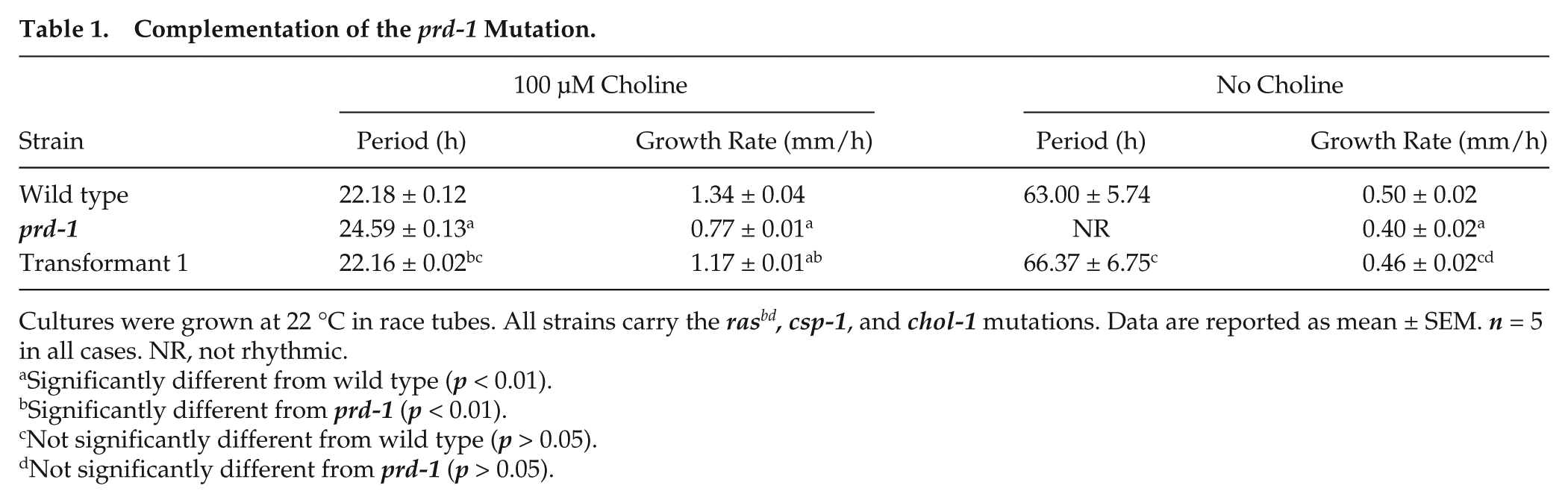
Cultures were grown at 22 °C in race tubes. All strains carry the rasbd, csp-1, and chol-1 mutations. Data are reported as mean ± SEM. n = 5 in all cases. NR, not rhythmic.
Significantly different from wild type (p < 0.01).
Significantly different from prd-1 (p < 0.01).
Not significantly different from wild type (p > 0.05).
Not significantly different from prd-1 (p > 0.05).
Analysis of the prd-1 Gene Sequence and Mutation
The transcript of NCU07839 is predicted to contain 4 introns (Figure 3A). The mutation identified in our sequencing of the prd-1 mutant is a single base pair change (G>A) at the first base of the third intron (Figure 3A and Suppl. Figure S1A). The 5′ splice site (ss) of this intron is GTAAGT, identical to the most common 5′ss found in a survey of N. crassa splicing signals (Collins et al., 2015). The prd-1 mutation changes the first base of this conserved 5′ss and therefore could affect the recognition of this intron by the splicing machinery.
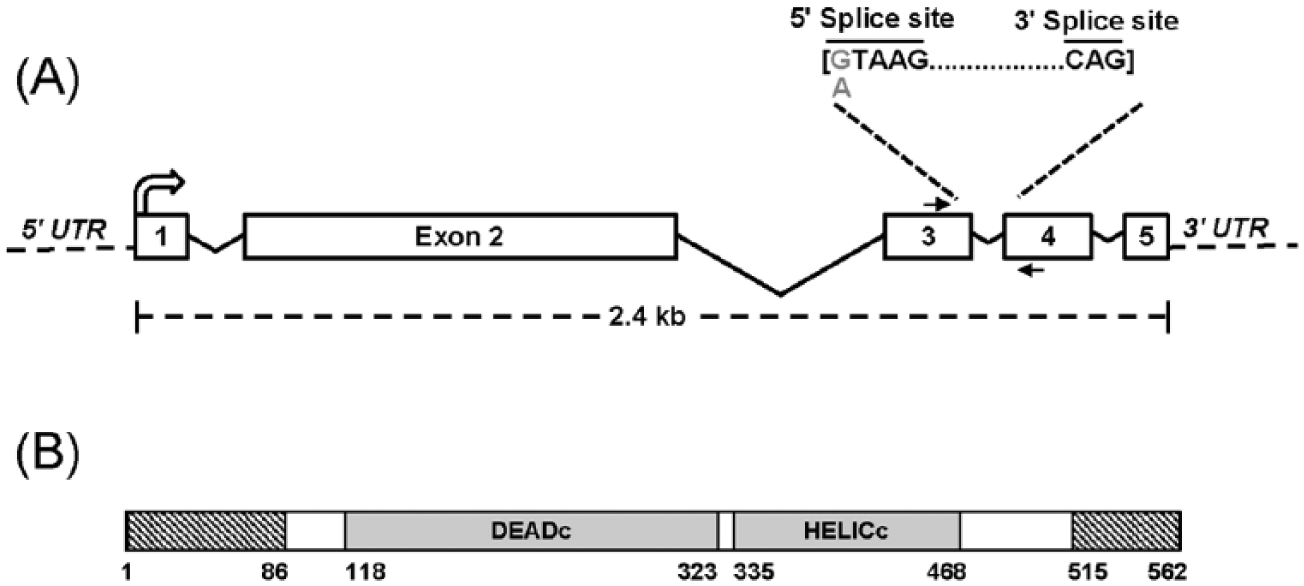
Organization of the prd-1 gene and protein domains. (A) The prd-1 gene (NCU07839; 2466 bp) contains 4 introns (sizes: 138 bp, 520 bp, 63 bp, and 57 bp). The prd-1 mutation is a single base substitution in the first nucleotide of the conserved 5′ splice site of the third intron (G to A). The primers highlighted with arrows were used to amplify complementary DNA from wild type or prd-1 mutant (see Figure 4) to demonstrate abnormal splicing in the prd-1 mutant. (B) Domain organization of the PRD-1 protein, showing conserved DEADc and HELICc domains found in RNA helicases. The numbers indicate position of the amino acid residues. PRD-1 also contains disordered regions (indicated in hashed boxes) in the amino- and carboxy-terminal regions.
The effect of the prd-1 mutation on messenger RNA (mRNA) splicing was investigated by isolating RNA, reverse transcribing it to cDNA, and amplifying a region containing the third intron using PCR primers in exons 3 and 4 (Figure 3A and Suppl. Figure S1A). RNA was isolated from cultures of wild type and mutant grown at both 22 °C and 30 °C to determine whether temperature affected splicing. As shown in Figure 4A, at both temperatures, the wild-type transcript was completely spliced (indicated by the band at about 200 bp), while the mutant also showed some unspliced transcript (at about 250 bp) as well as larger products. The PCR products from wild type and mutant were cloned into a blunt-end cloning vector and several clones were sequenced. All 5 of the wild-type clones that were sequenced gave the correctly spliced transcript, as expected (Figure 4B, line 2).
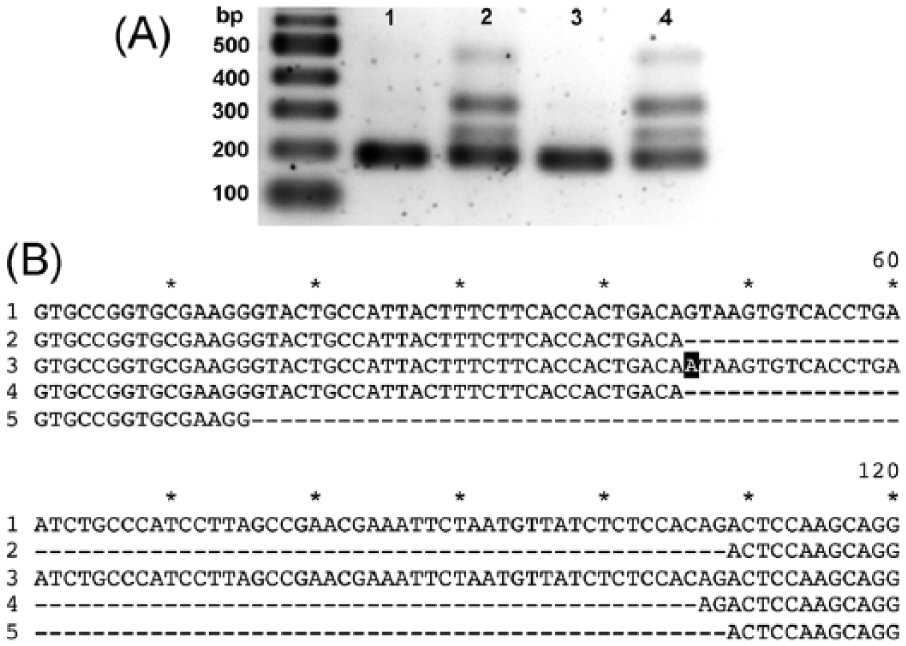
Effects of the prd-1 mutation on the splicing of NCU07839. (A) Agarose gel electrophoresis of polymerase chain reaction (PCR) products from complementary DNA (cDNA) of wild type (lanes 1 and 3) and prd-1 mutant (lanes 2 and 4). Cultures were grown at 22 °C (lanes 1 and 2) and 30 °C (lanes 3 and 4). The expected size of the correctly spliced PCR product is 192 bp. Molecular weight markers are indicated on the left. (B) Sequence alignments of cDNA of wild type and prd-1 mutant. The PCR products from cDNA templates shown in (A) were cloned into the pJET 1.2 vector and sequenced. In total, 120 nucleotides around the splice site are shown. Line 1: wild type unspliced (from genome sequence). Line 2: wild-type cDNA correctly spliced. Line 3: mutant cDNA unspliced, with mutation highlighted in black. Line 4: mutant cDNA incorrectly spliced with 2 additional nucleotides. Line 5: mutant cDNA incorrectly spliced with deleted nucleotides.
Eleven of the 15 mutant clones that were sequenced were found to be incorrectly spliced with 2 additional bases inserted at the point of splicing (Figure 4B, line 4). The wild-type cDNA sequence indicates that the 5′ and 3′ ends of the intron are GT-AG (Figure 4B, line 2), as predicted for the most common type of splice site in eukaryotes in general (Kupfer et al., 2004) and in N. crassa in particular (Collins et al., 2015). Two possible splice sites in the mutant could give the observed sequence, using either AT-AC or TA-CA (Figure 4B, line 4). The first of these corresponds to the “atac” site recognized by the minor U12 spliceosome found in higher eukaryotes (Turunen et al., 2013). However, although RNA genes for the major U2 spliceosome RNAs were found in N. crassa, no genes for the minor U12 spliceosome RNAs were found in N. crassa or any other ascomycota studied (López et al., 2008). In a survey of annotated splice sites in the N. crassa genome (Collins et al., 2015), a few AT-AC splice sites were found, but there were no RNA-seq data to confirm their use, so these sites were dismissed as annotation errors. AT-AC splice sites have been found in another ascomycota, Aspergillus (Kupfer et al., 2004), at very low frequency. It is known that AT-AC splice sites can in rare cases be spliced by the major U2 spliceosome in other organisms (Lin et al., 2010). The N. crassa spliceosome has been described as “promiscuous” in that it is capable of splicing a wide variety of splice sites (Collins et al., 2015). Therefore the AT-AC site is the most likely splice site in the prd-1 mutant.
Of the 15 mutant clones sequenced, one was unspliced and included the G-to-A base substitution (Figure 4B, line 3). One clone was correctly spliced and probably used ATAAGT as the 5′ss and the wild-type CAG as 3′ss. Two clones were found to be incorrectly spliced with a deletion of 30 nucleotides (Figure 4B, line 5). The most likely explanation is that the splicing machinery has used the next available upstream GT as the 5′ss and the wild-type CAG as 3′ss. The presence upstream of the 3′ss of a canonical branchpoint sequence CTAAT, the third most common branchpoint found in annotated introns in N. crassa (Collins et al., 2015), may provide additional information to direct the spliceosome to these sites in the mutant.
The NCU07839 ORF has been annotated in the N. crassa genome database (FungiDB, http://fungidb.org/fungidb; Stajich et al., 2012) as an adenosine triphosphate (ATP)–dependent RNA helicase dbp-2. The protein is predicted to contain a conserved DEAD/DEAH-box helicase domain, which includes the ATP-binding site, and a helicase conserved C-terminal domain (Figure 3B and Suppl. Figure S3). Analysis of the sequence using Phyre2 software (Kelley and Sternberg, 2009) predicts low-complexity disordered regions at the N- and C-termini of the protein (Figure 3B and Suppl. Figure S3).
These results indicate that the prd-1 mutation disrupts normal splicing of the third intron of the NCU07839 ORF. For the unspliced mutant transcript, translation is predicted to read through into the third intron, where a stop codon would produce a truncated protein (Suppl. Figure S3, line 2). The incorrectly spliced mutant transcript with an insertion of 2 bases would change the reading frame and is predicted to produce a longer protein before encountering a stop codon (Suppl. Figure S3, line 3). The incorrectly spliced transcript with a deletion would not change the reading frame but would change 1 amino acid and delete the following 10 (Suppl. Figure S3, line 4). In all cases, the mutation would alter the low-complexity C-terminal region but is predicted not to affect either the DEAD-box domain or the conserved C-terminal helicase domain. This low-complexity C-terminal region may mediate interactions with other proteins that are essential for PRD-1 function.
The Wild-Type prd-1 Gene Product Is Essential for Viability
The splicing defect in the prd-1 mutant described above may result in partially functional proteins or may be equivalent to a null mutation. The deletion of the NCU07839 gene produced by the Neurospora Knockout Project (Colot et al., 2006) is available from the FGSC as a heterokaryon, not a homokaryon, suggesting that the homokaryon may be lethal. We attempted to extract viable homokaryons from the heterokaryon using hygromycin selection. The knockout method is a gene replacement using a hygromycin resistance cassette (Colot et al., 2006), and therefore knockouts can survive on hygromycin-containing media. We purified uninucleate microconidia from the heterokaryon and plated them on hygromycin-containing media. In control experiments, a heterokaryon between an RFP-labeled strain and a GFP-labeled strain carrying a knockout of an unrelated gene produced many colonies on the selective media, almost all GFPs with a few red/green heterokaryons. Plating microconidia from the NCU07839 knockout heterokaryon strain, in contrast, produced only 2 colonies on selective media. Genotypes were determined by PCR, and the 2 colonies on selective media were heterokaryons. This failure to isolate viable homokaryotic knockouts indicates that the prd-1 gene product is essential for viability.
PRD-1 Is a Nuclear Protein
To determine the subcellular localization of PRD-1, a GFP fusion vector was constructed and transformed into a prd-1 mutant strain also carrying the chol-1 mutation. The transformants on high choline have growth rates similar to prd-1 wild type (Suppl. Figure S4), and on zero choline, the transformants show more sustained and clear banding compared to the prd-1 mutant (Suppl. Figure S4). These results indicate that the PRD-1–GFP fusion is functional.
The GFP-expressing transformants were observed under fluorescence microscopy along with a strain expressing a histone-H1-RFP fusion as a control. Figure 5A-D shows similar distributions of fluorescence for the 2 strains, indicating that the PRD-1 protein is found in the nucleus. Figure 5E is a close-up of the PRD-1–GFP labeled nuclei showing brightly labeled regions within the nuclei. Nearly all nuclei showed a similar distribution of GFP.
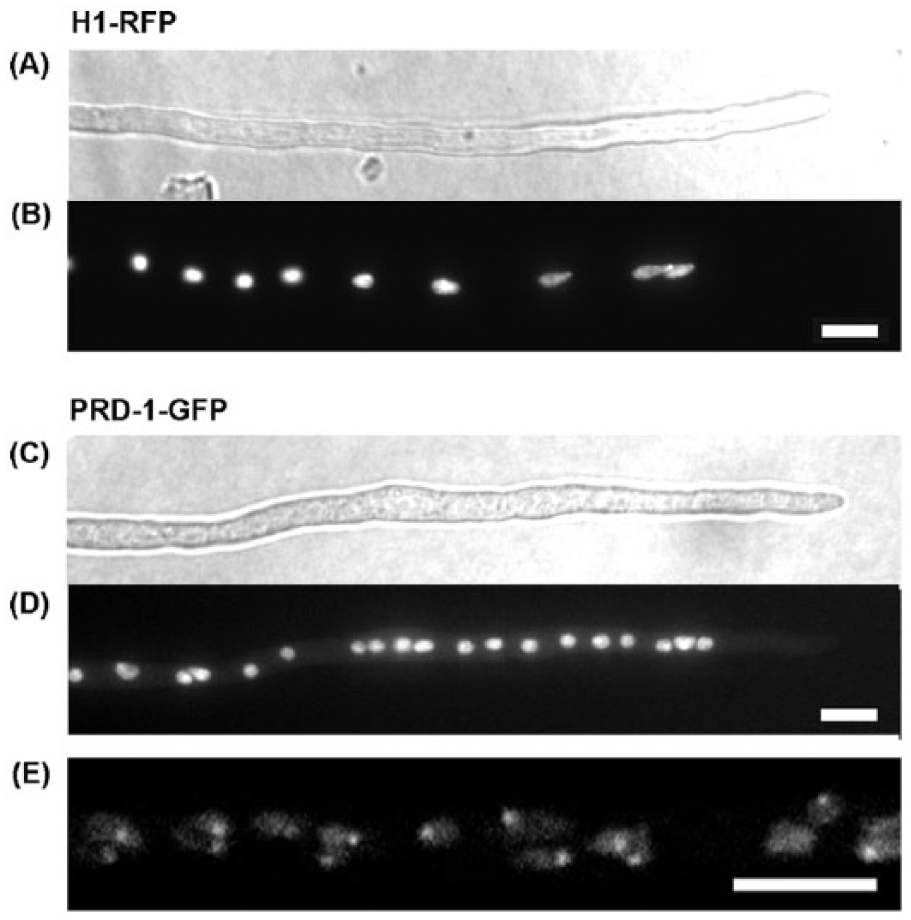
Nuclear localization of the PRD-1 protein. (A, B) RFP-tagged histone H1 was expressed in a prd-1+ strain (csp-1 his-3+::Pccg-1::hH1-RFP; chol-1 rasbd; inl). (C-E) PRD-1–GFP was expressed to complement a prd-1 mutant (csp-1 his-3+::Pccg-1::prd-1+-GFP; prd-1; chol-1 rasbd). (A, C) Young hyphae were observed by differential interference contrast imaging. (B, D) The same hyphae under fluorescence. The localization of H1-RFP and PRD-1–GFP was monitored in 100 hyphal tips and were found to localize in nuclei in all the tips (~1000 nuclei each). (E) Magnification of nuclei using confocal microscopy. Two consecutive Z-sections of 1 µm were merged. All scale bars are 5 µm.
Other RNA Helicases in N. crassa Have Minor Effects on FRQ-Less Rhythms
The results presented above demonstrate that PRD-1 is a member of the DEAD-box RNA helicase family. Another member of the DEAD-box RNA helicase family, FRH, is known to be involved in regulating the FRQ/WCC oscillator in the circadian system of N. crassa (as described in the Discussion). This raises the possibility that the effects of the prd-1 mutation on the circadian system may be due to a general pleiotropic effect of any mutation that disrupts RNA metabolism. If so, then other DEAD-box RNA helicases may also affect FRQ-less rhythmicity. We chose to assay the clock phenotypes of two, prd-6 and msp-8.
The msp-8 gene is identified as a pre-mRNA splicing factor and a DEAD/DEAH-box helicase (FungiDB, http://fungidb.org/fungidb; Stajich et al., 2012), and it maps to the same chromosomal region as prd-2 (Gardner and Feldman, 1981), which we have previously shown affects FRQ-less rhythms (Li and Lakin-Thomas, 2010). We therefore sequenced the msp-8 gene in the prd-2 mutant but did not find any sequence differences in comparison with the wild-type genome database (data not shown). Therefore we concluded that prd-2 is not msp-8.
Another prd mutation, prd-6, shortens the period of the conidiation rhythm in frq+ strains in a temperature-dependent manner (Morgan and Feldman, 1997) and is a null mutation (Compton, 2003). The prd-6 mutation shortens the period of the FRQ-less rhythm on geraniol medium (Lombardi et al., 2007). The PRD-6 protein is important in mRNA degradation (Zhang and Sachs, 2015).
To determine whether the prd-6 and msp-8 helicases affect FRQ-less rhythmicity in our assays, we constructed csp-1; chol-1 rasbd strains carrying these mutations, with and without the null frq9 mutation. In frq+ strains on high choline, we confirmed the previously reported period-shortening effect of prd-6 but did not find any significant effect of msp-8 on the period (Suppl. Table S5).
To assay the effects of these mutations on FRQ-less rhythmicity, we used 2 different assay methods. The strains used for these assays carried the chol-1 mutation to allow us to assay the FRQ-less rhythm in choline-depleted cultures. When chol-1 strains are supplemented with choline, the chol-1 defect is repaired and the cultures are indistinguishable from chol+ strains, allowing us to assay heat pulse entrainment behavior in the same genotypes.
We first grew the strains without choline in free-running conditions (constant darkness, 22 °C) to assay the FRQ-less rhythm seen in choline-depleted cultures (Lakin-Thomas and Brody, 2000). Because the effects of prd-6 were reported to be temperature sensitive (Morgan and Feldman, 1997), we also grew this strain at 25 °C. The essential findings were that neither mutation had a significant effect on the period of the rhythm in frqnull strains (Suppl. Table S5).
Our second assay for the FRQ-less oscillator used the heat-pulse entrainment protocol previously used to demonstrate the existence of an entrainable FRQ-less oscillator in frqnull strains (Merrow et al., 1999; Roenneberg et al., 2005; Lakin-Thomas, 2006a). In our protocol, cultures were grown on high choline in constant darkness at 22 °C and subjected to heat pulses (2 h at 32 °C) at regular intervals, using 3 different T cycles to entrain the conidiation rhythm. As seen in Supplemental Figure S5, neither msp-8 nor prd-6 had significant effects on entrainment of frqnull strains to the heat pulse protocol, unlike prd-1. We conclude that neither of these mutations has major effects on either of the FRQ-less rhythms we assayed, and they do not phenocopy the prd-1 mutation.
Discussion
The prd-1 mutation affects the period of the conidiation rhythm in frq wild type strains (Feldman and Atkinson, 1978) and has been shown to disrupt FLO-driven rhythms in frqnull strains in four different conditions (Lakin-Thomas and Brody, 1985; Lombardi et al., 2007; Li and Lakin-Thomas, 2010). Here we demonstrate that prd-1 also disrupts rhythms in White Collar-1–deficient strains. The simplest model for the circadian system (Li and Lakin-Thomas, 2010; Lakin-Thomas et al., 2011) that can account for these results is one in which there is only one oscillator that is observed under our various assay conditions when the FRQ/WCC TTFL is disrupted, and that oscillator (FLO) requires PRD-1. It is also possible to explain these results with more complex models in which a number of oscillators each drive independent output pathways, and the PRD-1 gene product has independent functions in each oscillator, but in the absence of evidence to differentiate between competing models, we prefer the simplest model.
We have mapped the prd-1 mutation and identified it as a single base substitution (G-to-A) in the conserved 5′-spliceosome recognition sequence of the third intron of this gene. We have identified the product of the prd-1 gene as a member of the DEAD-box RNA helicase family and demonstrated that the mutation affects splicing of the pre-mRNA, likely resulting in proteins with an altered C-terminal region, either due to truncation or altered amino acid sequence. Although the C-terminal region falls outside the conserved DEAD-box domain or the ATPase domain, it may be important for mediating protein-protein interactions that are vital for the function of PRD-1 in circadian rhythms and other cellular processes.
The DEAD-box RNA helicase family of proteins is involved in many aspects of RNA metabolism, including transcription, splicing, nuclear export, ribosome biogenesis, translation, and RNA stability (Linder and Jankowsky, 2011). These proteins can also function as RNA chaperones, promoting the folding of RNAs and assembly of ribonucleoprotein complexes (Jarmoskaite and Russell, 2014). Protein sequence similarity by PBLAST suggests that PRD-1 is homologous to the Dbp-2 DEAD-box RNA helicase in Saccharomyces cerevisiae (65% identity) and the DDX17 (66% identity) and DDX5 (59% identity) DEAD-box RNA helicases in mammals (Stajich et al., 2012). In yeast, Dbp-2 protein functions as an RNA chaperone associated with chromatin and regulates transcriptional fidelity (Cloutier et al., 2012). In mammals, p68 (DDX5) is involved in ribosome biogenesis, mRNA splicing, and microRNA processing. It can also function as a transcriptional coactivator, and this function does not always require the helicase catalytic activity (Janknecht, 2010). Both Dbp-2 and DDX5 are localized to the nucleus. We found (Figure 5) that PRD-1–GFP is localized to the nucleus and forms bright foci, similar to the localization of Dbp2-GFP to ribosomal DNA in the nucleolus of yeast (Cloutier et al., 2012) and reminiscent of the distribution of the heterochromatin protein HP1-GFP in the nucleus of N. crassa (Freitag et al., 2004a). The apparent localization of PRD-1 to the nucleolus, based on analogy with yeast, suggests that it may be involved in RNA processing and perhaps ribosome biogenesis.
The prd-1 mutant has a slow-growth phenotype (Feldman and Atkinson, 1978). The failure to isolate a homokaryon from the heterokaryotic knockout of this gene indicates that the PRD-1 protein is essential to viability of N. crassa. In yeast, Dbp-2 null mutants grow very slowly, and in mammals, DDX5 is an essential gene (Janknecht, 2010). The slow-growth phenotype of the prd-1 mutant in N. crassa may indicate that effects of the prd-1 mutation on the circadian system are pleiotropic effects of disruption of some essential aspect of RNA metabolism, or prd-1 may play a specific role in the clock mechanism and may have other roles in essential processes, or alternatively it may be that proper regulation of the circadian system (including the FLO(s)) depends on essential cellular processes. (Our results with prd-6 and msp-8 deletions indicate that disrupting the activity of an RNA helicase does not always affect FRQ-less rhythmicity, but these are not essential genes.)
FRQ-interacting RNA helicase (FRH) was found to be associated with FRQ protein by coimmunoprecipitation (Cheng et al., 2005). This protein is a DEAD-box helicase and appears to mediate the association between FRQ and WCC proteins (Cheng et al., 2005). The gene is essential, but when it is knocked down by RNA interference (Cheng et al., 2005) or in a partial-loss-of-function mutant (Shi et al., 2010), cultures become arrhythmic for conidiation. Heterokaryons of wild type with the partial-loss-of-function mutant have weak rhythms with normal periods (Shi et al., 2010). The ATPase function of FRH is not required for its role as a chaperone-stabilizing FRQ against degradation (Hurley et al., 2013) but is required to regulate hyperphosphorylation of FRQ (Lauinger et al., 2014). It remains to be determined whether the catalytic activity of PRD-1 is required for its function in regulating FLO(s) or if it serves as a scaffold to nucleate unknown protein complexes that are required for FLO(s).
RNA helicases have also been implicated in circadian rhythms in other organisms. In Drosophila, a mutation in a DEAD-box RNA helicase dprp43 was found to lengthen the circadian period, and the protein may be involved in splicing (Martinek, 2001). In mouse, the clock proteins PER and CRY form a complex to repress transcription, and this complex includes the RNA helicases DDX5 (orthologous to N. crassa prd-1), DHX9, and SETX (Padmanabhan et al., 2012).
These results focus attention on prd-1 as a member of the RNA helicase family specifically involved in regulating rhythmicity. Identifying the function(s) disrupted in the prd-1 mutant may lead to identification of cellular processes essential for the functioning of FRQ-less rhythmicity.
NOTE: The identification of prd-1 was first reported by our laboratory in 2013 (Firoozi, 2013). While the present article was in revision, a confirmation of our identification was reported by another group (Emerson et al., 2015).
Footnotes
Acknowledgements
Many thanks to Nardin Nano and Dieynaba Diallo for their contributions to the mapping of prd-1, to Stuart Brody for helpful discussions on RNA helicases, to Ron Pearlman for insights into RNA splicing, and to Ilhan Ali, Marian Guirguis, Amanda Mohabeer, Khalidha Nasiri, Advait Pandya, Di Wu, and Hajia Yakubu for technical assistance. Strains and plasmids were obtained from the Fungal Genetics Stock Center (Kansas City, MO).
Author Contributions
K.A. contributed to the data collection and analysis, experimental design and interpretation, supervision of research, and editing of the manuscript. G.F. contributed to the majority of data collection and analysis. K.M. contributed to data collection and analysis. P.L.L.-T. acquired funding; contributed to the overall direction of the research; contributed to experimental design and interpretation, data collection, and analysis; and wrote the first draft of the manuscript.
Conflict of Interest Statement
The author(s) have no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding provided by NSERC Discovery Grant 250133-2012.
Notes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.