
Abstract
The aim of this study was to evaluate the country-specific reporting status profile of immunomodulatory drugs (IMiDs)-related adverse events (ImrAEs) in real-world clinical practice, using data from the Japanese Adverse Drug Event Report (JADER) and Food and Drug Administration (FDA) Adverse Event Reporting System (FAERS) databases. Immunomodulatory drugs, including thalidomide and its derivatives, are a new class of anticancer and anti-inflammatory drugs. IMiD risk management programs have instituted sufficient measures to prevent fetal effects but do not address adverse effects experienced by patients themselves. To date, no study has compared ImrAE profiles across countries. Adverse events were defined using the preferred terms in the Medical Dictionary for Regulatory Activities. The number of reported adverse events related to IMiDs in each country (the United States and Japan) was investigated. In both Japan and the United States, myelosuppression, pneumonia, and neuropathy peripheral have been reported as adverse events suspected to be associated with IMiDs. Adverse event profiles differed between the countries. The number of adverse event reports for thalidomide increased transiently in the United States in 2008 following the multiple myeloma indication, and then exhibited a downward trend. The number of adverse event reports for lenalidomide and pomalidomide has increased in the United States since their launch. The number of transient reports increased in Japan in 2015, when pomalidomide was launched. In this study, the profile of ImrAEs was revealed using the FAERS and JADER databases. Our comparative safety study indicated the importance of comparing the safety profiles of IMiDs using post-marketing real-world data. It is important to focus on the adverse events experienced by patients taking IMiDs, as well as the effects of IMiDs on fetuses.
Keywords
Introduction
Immunomodulatory drugs (IMiDs) are a new class of anticancer and anti-inflammatory drugs that include thalidomide and its derivatives lenalidomide and pomalidomide. While thalidomide is marketed by Bristol-Myers Squibb as Thalidomide BMS® in Europe and THALOMID® in the United States (US), in Japan, Fujimoto Pharmaceutical Corporation markets thalidomide as THALED®. Lenalidomide and pomalidomide, developed by Celgene, are currently marketed by Bristol-Myers Squibb as Revlimid® and Pomalyst® in Japan and the US, and Revlimid® and Imnovid® in Europe.
Thalidomide, originally developed as a sedative drug, causes multiple adverse effects, including dysmelia (i.e. stunted limb growth) in humans, owing to its severe teratogenicity.1,2 Thalidomide has since been re-purposed for treating multiple myeloma, and derivatives such as lenalidomide and pomalidomide have been developed for treating blood cancer. 1
Thalidomide entered the US pharmaceutical market as a treatment for erythema nodosum leprosum (ENL) in 1998. It was subsequently approved as a treatment for multiple myeloma in 2006. In Europe, it was approved for the treatment of relapsed or refractory multiple myeloma in 2008. Meanwhile, in Japan, it was approved for the treatment of relapsed or refractory multiple myeloma in 2008, ENL in 2012, and Crow-Fukase syndrome (Polyneuropathy, Organomegaly, Endocrinopathy, M-Protein, and Skin Changes: POEMS syndrome) in 2021.
Lenalidomide entered the US pharmaceutical market in 2005 for the treatment of myelodysplastic syndromes (Figure 1). It was subsequently indicated for the treatment of multiple myeloma in 2006, mantle cell lymphoma in 2013, and relapsed or refractory follicular and marginal zone lymphomas in 2019. In Japan, lenalidomide is indicated for relapsed or refractory adult T-cell leukemia-lymphoma.

Schedule for the expansion of IMiD indications in the US, Japan, and EU.
Pomalidomide entered the US, European (2013), and Japanese (2015) pharmaceutical markets as a treatment for relapsed or refractory multiple myeloma. Pomalidomide was also indicated for the treatment of Kaposi sarcoma in the US in 2020.
Following the approval of thalidomide by the US Food and Drug Administration (FDA) in 1998, the manufacturer instituted a comprehensive program to control the prescription, dispensing, and use of the drug to ensure that fetal exposure to this teratogenic agent does not occur. This program is known as the System for Thalidomide Education and Prescribing Safety (S.T.E.P.S. [Celgene Corporation, Warren, NJ, USA]). 3
Since the 2007 FDA Amendments Act, the FDA has mandated sponsors of new drug applications, biologic license applications, and generic and biosimilar applications to submit a risk evaluation and mitigation strategy (REMS) program if the risks of the product cannot be addressed by routine product labeling. 4 As of 2015, more than 70 drugs, including thalidomide, have REMS programs. 5 To avoid embryo-fetal exposure, THALOMID® (thalidomide) is only available under a restricted distribution program called “THALOMID Risk Evaluation and Mitigation Strategy (REMS)®.” Only certified prescribers can prescribe THALOMID®, and only certified pharmacies can dispense THALOMID® in the THALOMID REMS® program. To receive THALOMID®, all patients must be enrolled in the THALOMID REMS® program and agree to comply with its requirements. 6 The thalidomide derivatives Revlimid® (lenalidomide) and Pomalyst® (pomalidomide) are also controlled by REMS.
In Japan, thalidomide was approved for the treatment of multiple myeloma under a risk management program (RMP), named TERMS®, in 2008 (Figures 1 and 2). 5 TERMS® is the first RMP in Japan, and similar to REMS in the US, it is a patient–participatory program. 5 TERMS® registers patients, physicians, pharmacists, and other related parties with the TERMS® Center, which checks for TERMS® compliance with each prescription and centrally manages all processes from distribution to medication administration. 5 In Japan, Revlimid® and Pomalyst® prescriptions are restricted by RevMate® to prevent fetal effects. Similarly, Europe and Australia have managed the appropriate use of IMiDs. In Germany, the Bundesinstitut für Arzneimittel und Medizinprodukte manages T-prescriptions, registers prescribing physicians, and provides patient education (Figure 2). 7
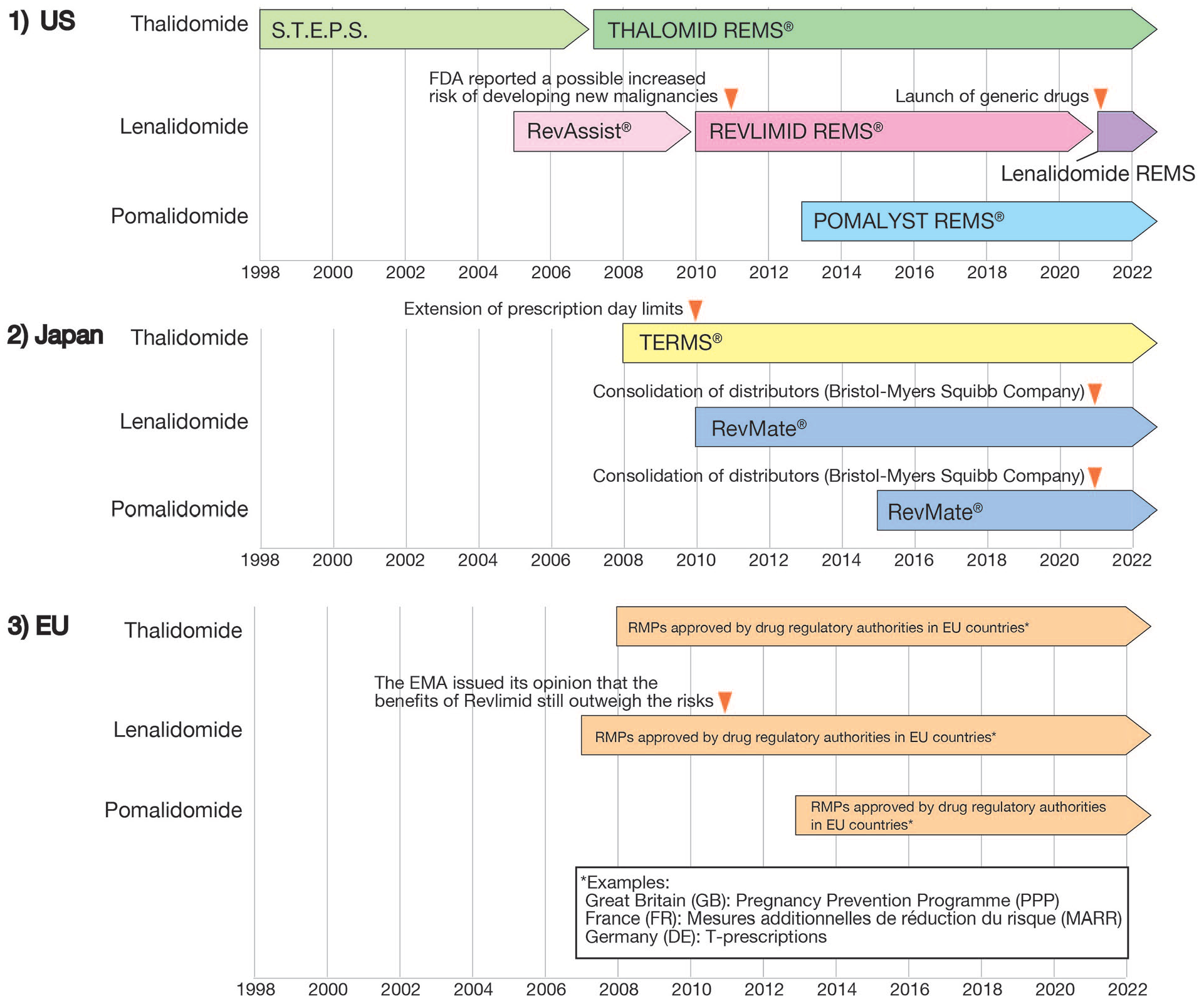
Launch schedule for IMiD RMP in the US, Japan, and EU.
IMiD RMPs have instituted sufficient measures to prevent fetal effects but do not address adverse effects experienced by patients themselves. Various adverse events are associated with IMiDs, such as myelosuppression, and effects on patients beyond fetal teratogenicity should be examined as soon as possible. To date, no study has compared IMiD-related adverse event (ImrAE) profiles across countries. In this study, we analyzed the occurrence of adverse events associated with IMiDs using the Spontaneous Reporting System (SRS) to evaluate the ImrAE profile in two countries (the US and Japan).
Materials and methods
Data sources
Two SRS data sources were used: (a) the FDA Adverse Event Reporting System (FAERS) database (www.fda.gov) and (b) the Japanese Adverse Drug Event Report (JADER) database (www.pmda.go.jp). All data from the FAERS and JADER databases were fully anonymized by the regulatory authorities before use. We built a relational database, which integrated the data tables, using FileMaker Pro version 20.3.2 (Claris International Inc., Santa Clara, CA, USA).
Since 1968, the FAERS database has accumulated reports from healthcare professionals, manufacturers, and consumers from the US as well as from other countries around the world. 8 The FAERS database comprises seven data tables: patient demographics and administrative information (DEMO), drug/biological information (DRUG), adverse events (REAC), patient outcomes (OUTC), report sources (RPSR), drug therapy start and end dates (THER), and indications for use or diagnosis (INDI). Drugs in the FAERS are classified into four categories: “primary suspect,” “secondary suspect,” “concomitant,” and “interacting” drugs, according to the anticipated degree of involvement in adverse events. Drugs assigned as primary suspected drugs were considered for analysis, covering reports from January 2004 to December 2021. The FDA’s recommendation to adopt the most recent case numbers to identify duplicate reports from the same patient was followed, and the duplicates were excluded from the analyses.
The publicly available JADER dataset, which includes information recorded between April 2004 and January 2022, was downloaded from the Pharmaceutical and Medical Devices Agency (PMDA) website (www.pmda.go.jp). The JADER database consists of four tables: (1) DEMO (patients’ demographic information); (2) DRUG (drug information); (3) REAC (adverse event information); and (4) HIST (primary disease information). In the DRUG table, each drug is assigned a code according to its association with adverse drug reactions, namely, “suspected drug,” “concomitant drug,” and “interacting drug.” The analysis was restricted to reports where drugs were recorded as a “suspected drug.”
Target drugs
The Anatomical Therapeutic Chemical (ATC) classification system described by the World Health Organization (WHO) Collaborating Centre for Drug Statistics Methodology was used to define drugs. Three IMiDs were selected: thalidomide (ATC code L04AX02), lenalidomide (ATC code L04AX04), and pomalidomide (ATC code L04AX06).
Breakdown of IMiD-related adverse events
We analyzed reports of ImrAEs in the US by searching only the US as the reporting country in the FAERS database. Japanese reports were examined using the JADER database, which includes more reports from Japan than the FAERS database. Adverse events were defined using preferred term (PT) in the Medical Dictionary for Regulatory Activities (MedDRA)/Japanese version 23.1 (MedDRA/J, https://www.jmo.pmrj.jp/). However, the words “death,” “disease progression,” “malignant neoplasm progression,” “multiple myeloma,” and “plasma cell myeloma” were considered as diseases treated with IMiDs and related patient conditions, therefore they were excluded from the summary table and noted in the footnotes.
Statistical analysis (Reporting odds ratio)
To ascertain adverse event signals, we calculated the reporting odds ratio (RORs), which was established using a disproportionality analysis. 9 To compare one of the index groups with the reference group, we calculated ROR values as (a × d)/(b × c) and expressed the data as point estimates with 95% confidence interval (CI). If the lower limit of the 95% CI of the ROR was greater than one, the ROR was considered an adverse event signal. 9 Two or more cases were required to positively identify such signals. 9
In the detection of ROR signals, calculations were first performed targeting “primary suspect” in FAERS and “suspected drugs” in JADER (Table 1). However, in real-world pharmacovigilance, it is necessary to investigate unknown relationships between adverse events and drugs. Therefore, in FAERS, the target reports were expanded to include not only “primary suspect” but also “secondary suspect,” “concomitant,” and “interaction” drugs. In JADER, the target reports were extended to “suspected drug” to include “concomitant drug” and “interacting drug,” and the ROR was calculated accordingly (Table 2).
Reporting odds ratio of adverse event reports related to immunomodulatory drugs limited by “primary suspect” for the FAERS database and “suspected drug” for the JADER database.
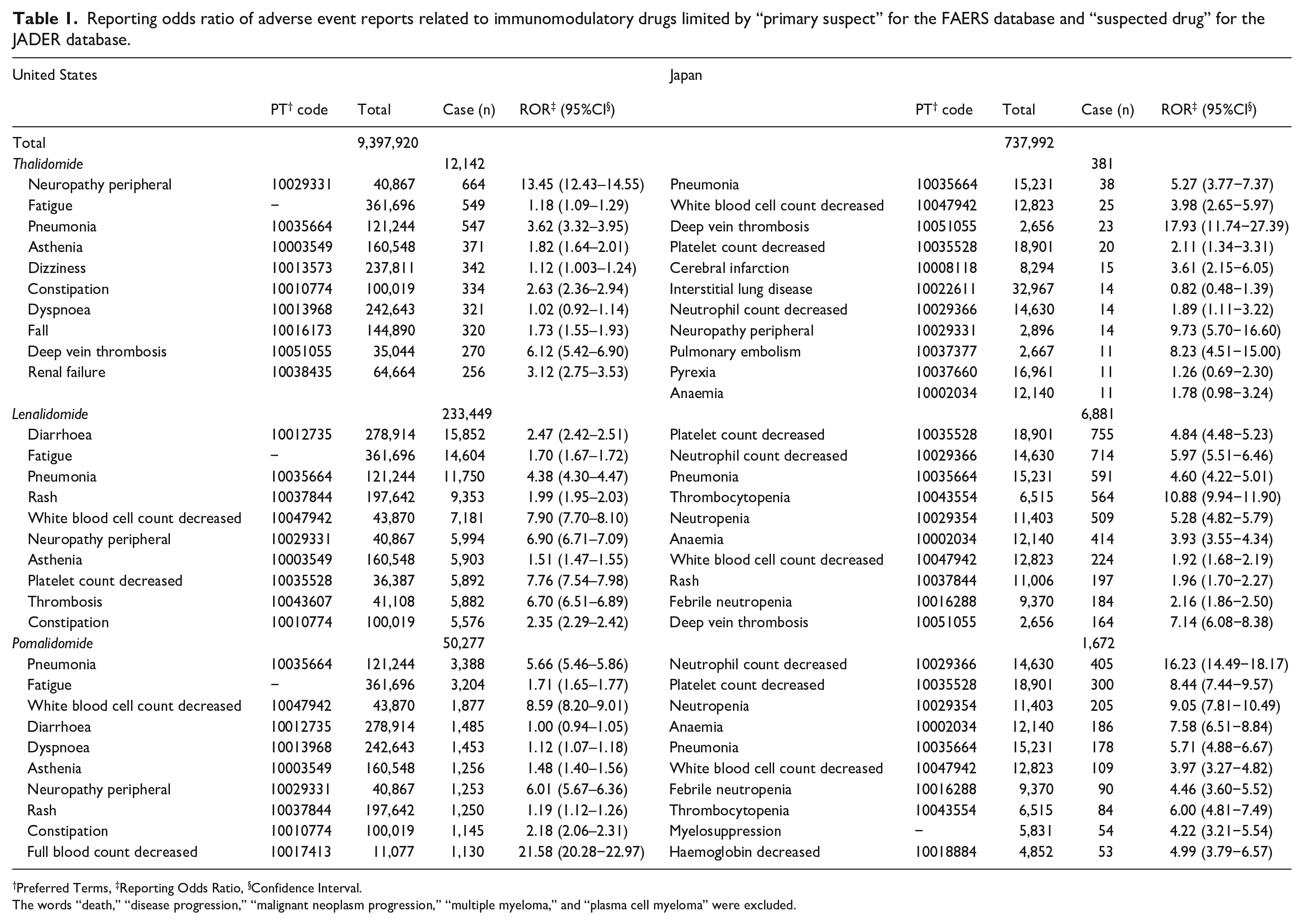
Preferred Terms, ‡Reporting Odds Ratio, §Confidence Interval.
The words “death,” “disease progression,” “malignant neoplasm progression,” “multiple myeloma,” and “plasma cell myeloma” were excluded.
Reporting odds ratio of adverse event reports related to immunomodulatory drugs in the US and Japan.
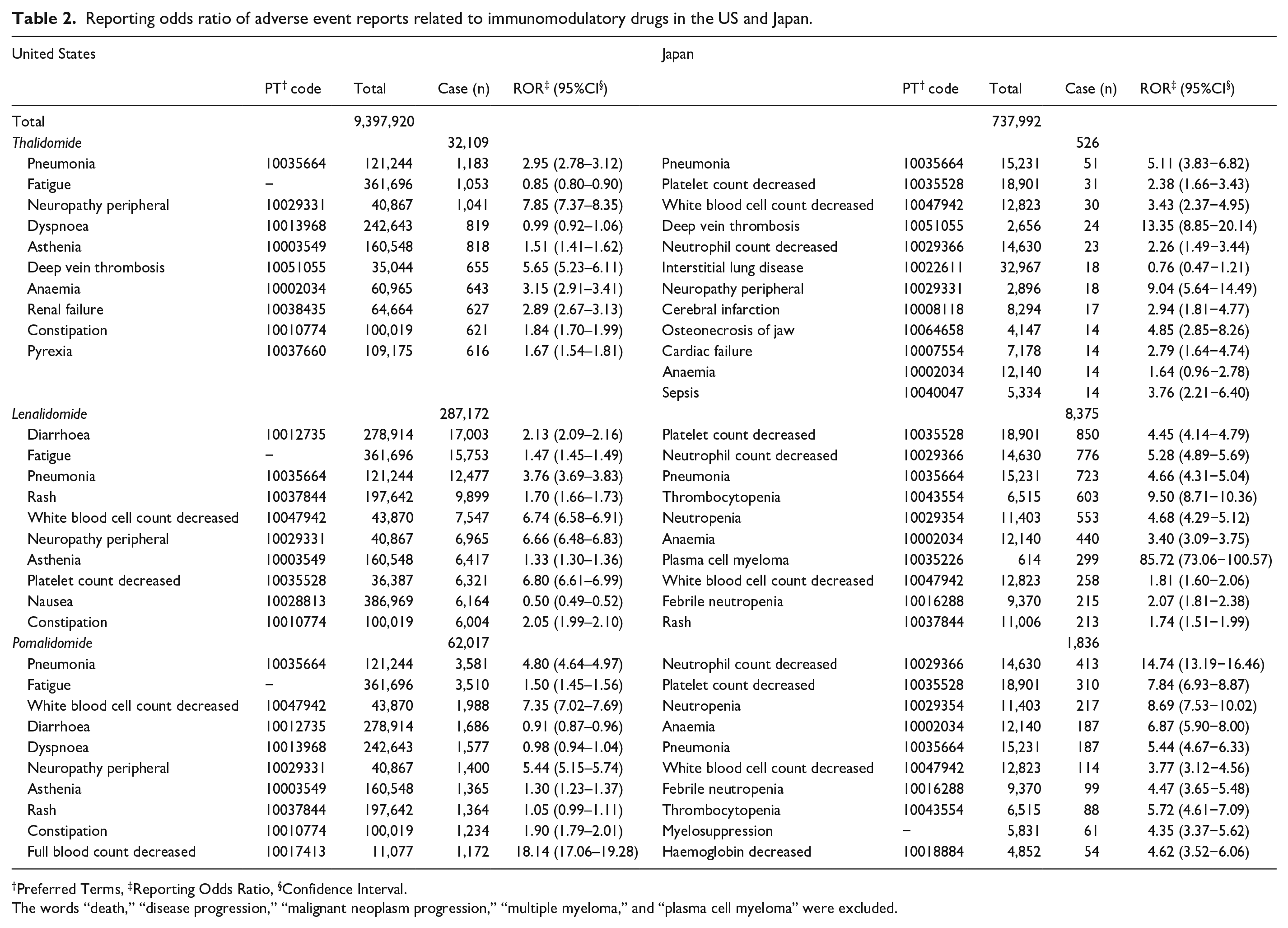
Preferred Terms, ‡Reporting Odds Ratio, §Confidence Interval.
The words “death,” “disease progression,” “malignant neoplasm progression,” “multiple myeloma,” and “plasma cell myeloma” were excluded.
Adverse event profiles for the US and Japan
The reporting ratio of ImrAEs was investigated by year. We calculated the percentage of ImrAE reports by dividing the number of ImrAE reports per country by the total number of adverse event reports per country. The association between the reporting year and events such as IMiD indication expansion and administrative intervention was investigated (Figures 1–3).
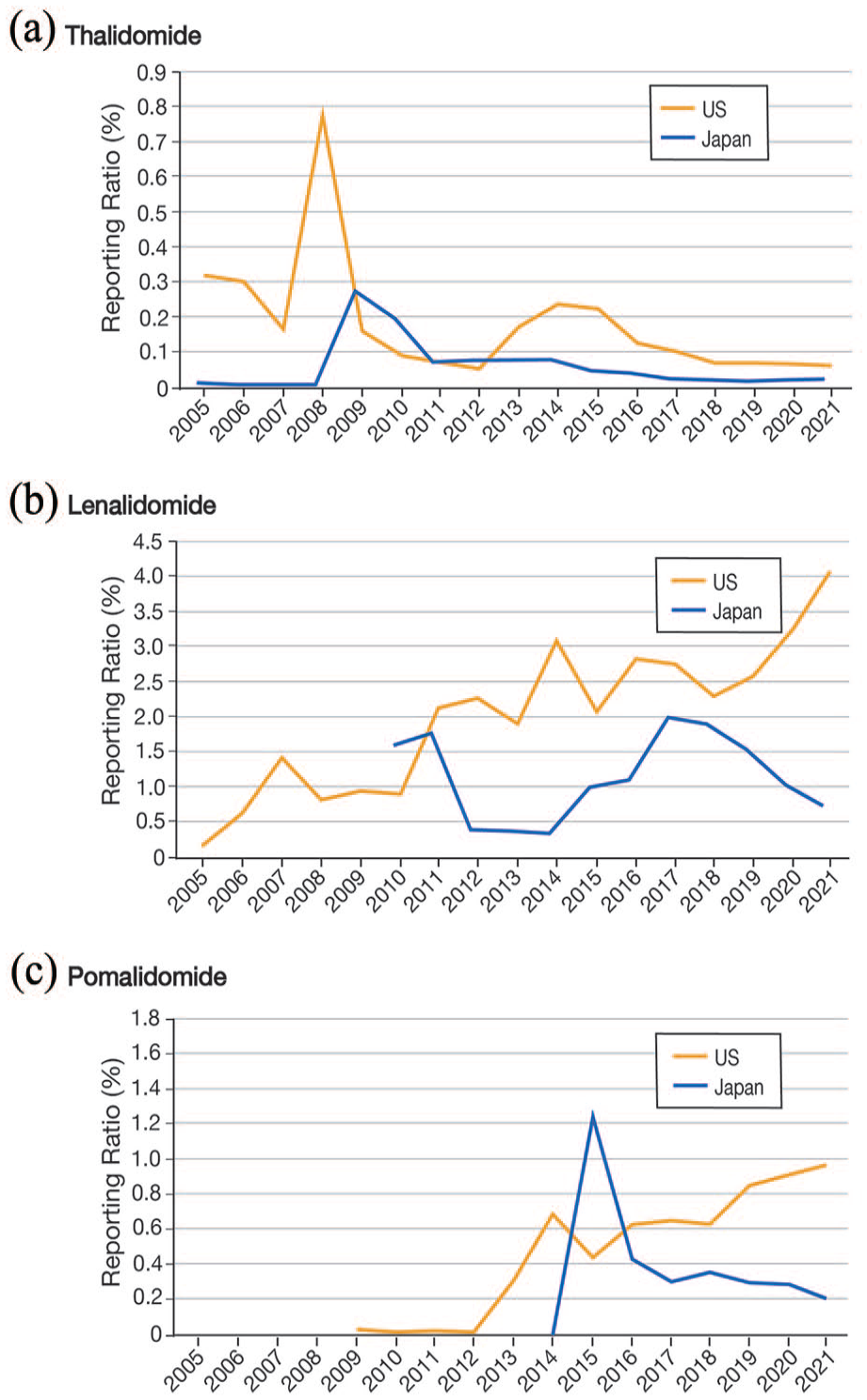
Percentage of IMiD-related adverse events (ImrAEs) reported in the US and Japan. The yellow line represents the US, and the blue line represents Japan.
Time-to-onset analysis
Recently, analysis of time-to-onset using the Weibull shape parameter (WSP) has been proposed to detect the signals for AEs by utilizing time-to-event data without requiring a reference population.10 –13 The WSP can describe the varying incidence of AEs and can evaluate hazard functions for detecting AEs. The clarification of the time-to-onset profile of ImrAEs will enable the provision of information on the timing of adverse event onset to those taking IMiDs. It will contribute to minimizing the damage from adverse events. To assess the time-to-onset profile, the median time from the first prescription of each drug to the onset of ImrAEs was used in conjunction with the interquartile range and WSP.10,14 In the FAERS database, it is difficult to match the date of adverse event onset with the start date of each drug, owing to the structure of the database. Therefore, in this study, the JADER database was used to analyze onset dates. The JADER database recorded multiple drugs administered for different durations in a single case and the occurrence of adverse events long after the start of IMiD treatment may have been influenced by concomitant medications. In other words, the occurrence of adverse events after a long period may not be related to IMiDs. Therefore, data more than 90 days after the start of treatment were excluded from the analysis. The WSP represents the failure rate distribution over time. A larger scale value (α) of the Weibull distribution indicates wider data distribution. A smaller scale value (α) shrinks the data distribution. The WSP (β) has been used to determine the level of hazard over time without a reference population. When β is equal to 1, the hazard is considered to be constant over time.
Statistical analyses were performed using JMP Pro 17 software (SAS Institute Inc., Cary, NC, USA).
Results
Breakdown of IMiD-related adverse events
The FDA Adverse Event Reporting System (FAERS) database contained 14,475,614 (US report: 9,397,920) reports that were submitted between January 2004 and December 2021. The Japanese Adverse Drug Event Report (JADER) database contained 737,992 reports that were submitted between April 2004 and January 2022. Regarding the FAERS data restricted to “primary suspect” and the JADER data restricted to “suspected drugs,” the number of adverse event reports related to IMiDs in the FAERS and JADER databases was 335,310 (US report: 295,868) and 8,934, respectively. The results of the descriptive analysis of basic patient information, including sex and age, are summarized in Supplemental Table S1. The number of adverse event reports for thalidomide in the US and Japan were 12,142 and 381; of lenalidomide, 233,449 and 6,881; and of pomalidomide, 50,277 and 1,672, respectively.
The number of reported cumulative cases of ImrAEs was examined by country, with the top 10 listed in Table 1. Regarding the FAERS data restricted to “primary suspect” and the JADER data restricted to “suspected drugs,” the RORs for the US and Japan are summarized in Table 1. In Japan, adverse events related to myelosuppression (e.g., neutrophil count decreased (PT code: 10029366) and platelet count decreased (PT code: 10035528)) and pneumonia (PT code: 10035664) were the most frequently reported events for the three drugs (Table 1). In the US, all three drugs ranked in the top 10 in terms of the number of reports of neuropathy peripheral (PT code: 10029331), fatigue, and pneumonia (PT code: 10035664) (Table 1). In Japan, there were no reports of spontaneous abortion or fetal malformations due to IMiDs. However, spontaneous abortions due to thalidomide and lenalidomide have been reported in the US (Table 3). When focusing on “primary suspect,” “secondary suspect,” “concomitant,” and “interacting” in FAERS, and on “suspected drug,” “concomitant drug,” and “interacting drug” in JADER, in Japan, adverse events related to pneumonia (PT code: 10035664) and myelosuppression (neutrophil count decreased (PT code: 10029366) and platelet count decreased (PT code: 10035528)) were the frequently reported events for the three drugs (Table 2). In the US, all three drugs ranked in the top 10 in terms of the number of reports of pneumonia (PT code: 10035664), fatigue, and myelosuppression (Table 2).
List of spontaneous abortion-related adverse event reports in the US.

Adverse event profiles for the US and Japan
In the US, thalidomide-related adverse events were most frequently reported in 2008 (0.77%) (Figure 3(a)). The reporting ratios have also increased from 2008 to 2009 in Japan. The reporting ratio of thalidomide-related adverse events in Japan was the highest in 2009 (0.28%). Since 2010, the number of reports has decreased, increasing slightly during 2012–2014.
The reporting ratio of lenalidomide-related adverse events was higher in the US than in Japan (Figure 3(b)). In the US, the reporting ratios increased significantly in 2007, 2011, and 2014. The reporting ratio of lenalidomide-related adverse events has gradually increased in the US, peaking at 4.06% in 2021. The reporting ratio of lenalidomide-related adverse events reported in the JADER database decreased significantly from 1.78% in 2011 to 0.38% in 2012 but increased in 2015, reaching a maximum of 1.96% in 2017.
The US and Japan had a high reporting ratio of adverse events for pomalidomide from 2014 to 2015 (Figure 3(c)). The reporting ratio of pomalidomide-related adverse events in the US has been increasing in recent years, reaching 0.98% in 2021. The reporting ratio of pomalidomide-related adverse events in the JADER dataset was the highest in 2015 (1.22%), decreasing thereafter.
Time-to-onset analysis
Figure 4 (1) and 4 (2) show a histogram of the number of ImrAE cases from day 0 to day 90 in the JADER database. The median number of days to onset for most adverse events was 30 days or less. Pneumonia and deep vein thrombosis have often been reported after 30 days. The median and WSP for lenalidomide and pomalidomide were similar for most adverse events.
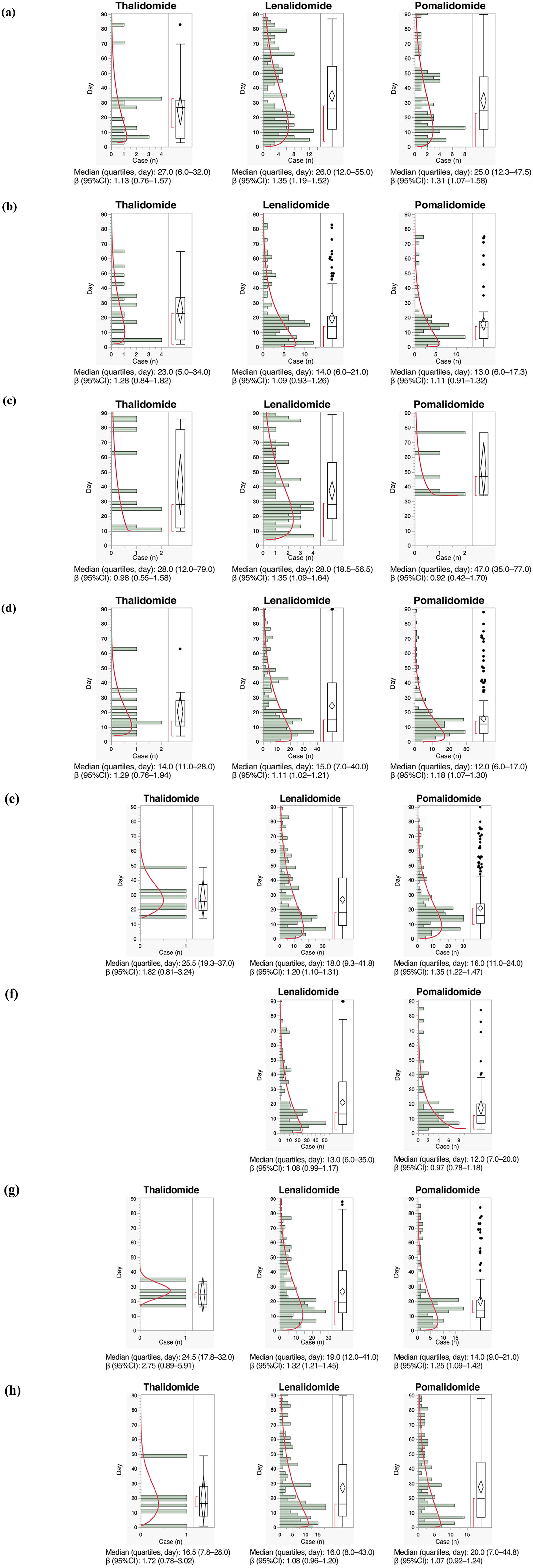
Box plot and Weibull shape parameter (β) of ImrAEs: (a) Pneumonia, (b) White blood cell count decreased, (c) Deep vein thrombosis, (d) Platelet count decreased, (e) Neutrophil count decreased, (f) Thrombocytopenia, (g) Neutropenia, and (h) Anaemia. Histogram and Weibull shape parameter of ImrAE for each drug in the ATC classification. Right panel shows box plots, which represent the median (the horizontal line within the box). The ends of the box represent the 25th and 75th quantiles, also expressed as the 1st and 3rd quartile, respectively. The confidence diamond contains the mean and the upper and lower 95% CIs of the mean. The whiskers extend to the outermost data point that falls within the distances of 1.5 times the length of the inner quartiles. The bracket outside the box indicates the shortest half, which is the densest 50% of the observations. Abbreviation: 95%CI, 95% confidence interval.
Discussion
Breakdown of IMiD-related adverse events
Adverse events related to myelosuppression (e.g., neutrophil count decreased and platelet count decreased) and pneumonia were the frequently reported ImrAEs in Japan. Although the complete pathogenesis of IMiD-induced myelosuppression is not yet clear, the involvement of the transcription factor PU.1 has been suggested. 15 In Japan (JADER) and the US, adverse event reports were related to myelosuppression.
The Japanese package insert for IMiDs lists myelosuppression-related adverse events such as neutropenia, thrombocytopenia, and anemia, as well as pneumonia and peripheral neuropathy, as serious side effects.16 –18 The adverse events listed in the Japanese package insert for IMiDs and the results of this study are generally consistent.
ImrAE reports in the US were generally consistent with the adverse events listed in the US package insert for IMiDs. The US package insert also cautions about myelosuppression-related adverse events, as in Japan.19 –21 Although the US package insert does not list pneumonia as a common adverse event,19 –21 we found that pneumonia was frequently reported for all three drugs (Table 1). There is a lack of information on the mechanism of pneumonia caused by IMiDs. The Japanese package insert cautions about pneumonia resulting from infection,16 –18 and healthcare providers should be aware of pneumonia in patients taking IMiDs. Anticancer drugs commonly cause hematologic toxicity, including myelosuppression and hematopoiesis. Therefore, these adverse effects may be less frequently reported to the FAERS and JADER, especially minor ones, because they are not considered serious. It would be desirable to consider the severity grade of adverse events, but such information is rarely entered in the JADER. This could be accomplished with more detailed retrospective studies using electronic medical record records.
In Table 2, which is not limited to suspected drugs, osteonecrosis of the jaw (PT code: 10064658) was ranked among the top 10 thalidomide-related reports in Japan. However, all 14 reports on osteonecrosis of the jaw listed bisphosphonates or receptor activator of nuclear factor-κB ligand (RANKL) inhibitors as the suspected drugs. Bisphosphonates and RANKL inhibitors are representative drugs associated with osteonecrosis of the jaw. 22 These drugs are considered more likely to contribute to the pathogenesis of osteonecrosis of the jaw than thalidomide. Caution should be exercised when including “concomitant” and “secondary suspect” drugs in the analysis, as this may lead to the identification of associations between less relevant drugs and adverse events.
“Death,” “disease progression,” and “malignant neoplasm progression” were considered outcomes, whereas “multiple myeloma” and “plasma cell myeloma” were regarded as primary diseases. Therefore, in this study, these terms were not treated as ImrAEs and were excluded from the analysis. Several clinical trials of lenalidomide have demonstrated a higher incidence of malignancy in the lenalidomide group compared to the control group.23 –25 In several clinical trials for multiple myeloma, deaths have been reported in the IMiD-containing groups owing to treatment-emergent adverse events.26 –28 The US package insert for Revlimid® mentions increased mortality in chronic lymphocytic leukemia and mantle cell lymphoma in addition to fatal liver failure and fatal tumor lysis syndrome in the WARNINGS AND PRECAUTIONS section. 20 Therefore, IMiDs and death might be associated with malignancy appearance. Meanwhile, the SRS is a database of actual adverse event reports; however, minor adverse events are often overlooked and might be underreported compared to serious ones. As a result, serious terms such as “death” and “disease progression” are likely to be reported more frequently. This trend varies by reporting country and reporting author. For example, in the case of thalidomide in Table 1, the number of death reports from the United States is 1,039, whereas in Japan, it is 3 (data not shown). The occurrence of death and malignancies in patients taking IMiDs is influenced by various patient backgrounds, and reports from the SRS alone cannot clarify the causal relationship between IMiDs and these outcomes. For these reasons, we avoid further analysis of the relationship between IMiDs and “death” or “disease progression” using the SRS in this study.
The types of reports registered in the SRS databases differ between Japan and the US. The JADER database is limited to individual case reports, whereas the FAERS database includes periodic case reports. Nomura et al. compared the differences in adverse event reporting between the JADER and FAERS databases. 29 They showed distinct differences in the reported drugs, adverse events, and event severity, as well as in the average number of reported events per case. 29 The two databases exhibit different characteristics, which may arise from variations in reporting rules and practices in each country, such as reporters and reported adverse event terms. 29 When coding adverse events in MedDRA, there is a tendency to exclude non-serious events in individual case reports in Japan. In contrast, non-serious adverse events are included in individual case reports in the US. 29 However, owing to the data structure of the JADER and FAERS databases, it was not possible to distinguish between serious and non-serious adverse events in individual cases. In addition, regional differences in drug prescriptions, diet, and medical environment can lead to differences in reporting. 30
Adverse event profiles for the US and Japan
For thalidomide, the reporting ratio in the US increased significantly in 2008. The change from S.T.E.P.S. to THALOMID REMS® in 2007 might be a contributing factor. Reporting ratios have increased from 2008 to 2009 in Japan. This is thought to be due to the launch of thalidomide. For example, thalidomide was approved in Japan in October 2008 for the treatment of relapsed or refractory multiple myeloma. With the addition of indications for ENL in May 2012 in Japan, there was an increase in the number of reports during 2012–2014.
The databases encounter many challenges, including underreporting, Weber effect, and notoriety bias. 31 The Weber effect refers to a substantial increase in spontaneous adverse drug reaction reporting, particularly during the first 2 years after initial drug approval, which then plateaus and eventually drops. 31 Notoriety bias is defined as “a selection bias in which a case has a greater chance of being reported if the subject is exposed to a factor known to cause, thought to cause, or likely to cause an event of interest.” 31 The transient increase and subsequent decrease in reporting ratios in each country with the addition of the thalidomide indication may be due to the Weber effect.
For lenalidomide, the reporting ratios in the US increased in 2007 and after 2011. Lenalidomide was approved for the treatment of myelodysplastic syndromes in December 2005, multiple myeloma in June 2006, and mantle cell lymphoma in June 2013, and the reporting ratios have increased accordingly. RevAssist® was transitioned to Revlimid REMS® in 2010. The fact that RevAssist®, which was an RMP for Revlimid® alone, is now managed under REMS, similar to other drugs requiring management, may have contributed to the increase in the number of reports. In Japan, lenalidomide was approved for the treatment of relapsed or refractory multiple myeloma in June 2010. Lenalidomide was approved for the treatment of untreated multiple myeloma in December 2015. Consequently, the number of adverse event reports increased between 2015 and 2016. In March 2017, lenalidomide was approved as an additional indication for relapsed or refractory adult T-cell leukemia-lymphoma, greatly expanding the number of patients eligible for lenalidomide therapy. Consequently, the number of adverse event reports increased through 2017. We did not consider the situation in Europe in this study. In Great Britain, the reporting ratio increased from 2011 to 2012. Great Britain did not expand the indication around 2011. The European Medicines Agency opines that the benefits of Revlimid® still outweigh the risks, 32 which may have influenced the increase in prescriptions and adverse drug reaction reports.
For pomalidomide, the reporting ratios increased in all countries as pomalidomide has recently entered the market. In the US, reports of adverse events associated with pomalidomide have increased in number in recent years. This may be partially because the drug was indicated for the treatment of Kaposi sarcoma in May 2020. In Japan, pomalidomide was approved for the treatment of relapsed or refractory multiple myeloma in May 2015. A three-drug combination of pomalidomide, bortezomib, and dexamethasone was approved in September 2018; however, the number of adverse event reports has not increased.
Jiang et al. calculated the percentage of adverse events reported for each IMiD from 2013 to 2021 using the FAERS. 33 The trend in the percentage reported by Jiang et al. is similar to the trend in the percentage of ImrAE reports from the US observed in this study. Jiang et al. reported that in the FAERS, approximately 90% of ImrAE reports were from the US. 33 This finding may be attributed to the fact that the majority of FAERS reports are from the US. We also compiled the trends in the percentage of thalidomide and lenalidomide reports from 2005 and compared them with the reporting situation in Japan.
Adverse events of IMiDs have been reported using various data sets.33 –39 Meta-analyses have suggested an increased risk of lenalidomide adverse effects and acute lymphocytic leukemia 34 and infection. 35 Using the Celgene Global Drug Safety database, the most common reason for lenalidomide discontinuation has been reported to be a non-serious rash. 36 There have been studies on lenalidomide-related thrombosis and embolism, and lung toxicity,37,38 and the association between pomalidomide and lung toxicity 39 using the JADER database. The number of reports or ROR results of our study show similar trends to these results.
Time-to-onset analysis
The median and WSP for lenalidomide and pomalidomide were similar for most adverse events. Pomalidomide is often used in heavily pretreated patients with highly compromised bone marrow function, which explains the higher and earlier incidence of neutropenia with lenalidomide, 40 but no significant difference was observed. The dosage of IMiDs was not included in the analysis in this study. Although it would be desirable to investigate the time-to-onset by dosage, this is difficult because the JADER does not have sufficient dosage information. To calculate the dosage of IMiDs, it is necessary to accurately understand the regimen, single dose, daily dose, and number of doses. However, JADER contains very little information about the regimen, and the actual input data often includes missing values or situations where the reporter may have confused the single dose with the daily dose. As a result, it is difficult to calculate the exact dosage. We have already analyzed the effects of herbal medicine dosage on interstitial pneumonia or metformin dosage on lactic acidosis using the dosage information from the JADER.41,42 It is apparent that the daily dose or single dose affects the occurrence of adverse events. However, to our knowledge, there is no widely accepted analysis method that combines dose and time-to-onset information in studies using SRS.
Risk management
Various measures have been taken in the RMP to prevent effects on fetus. However, deviations that may affect the fetus have been reported. Providing IMiDs to an unregistered patient is a serious incident that can have fetal effects in some cases. Failure to perform routine pregnancy tests has also been reported, which can lead to missed fetal exposure. In Japan, there have been no reports of misadministration of IMiDs to pregnant women or during pregnancy among those taking IMiDs. However, there have been cases of pregnant nurses touching Revlimid® with their bare hands; thus, education of patients as well as healthcare workers needs to be strengthened. 43
The administration of IMiDs to a pregnant woman or the failure of an IMiD user to use adequate contraception with a pregnant female partner constitutes deviation from the RMP. Analysis of the FAERS database revealed reports of spontaneous abortions in patients taking IMiDs and their partners in the US. Although it is difficult to confirm a direct causal relationship between spontaneous abortion and IMiD exposure and there is no doubt that the IMiD RMP to prevent fetal effects is being implemented with due diligence worldwide, a possible deviation from the RMP may be suggested.
Various adverse events have been reported following the use of IMiDs. It is as important to prevent adverse effects in patients as it is to prevent effects on the fetuses in order to continue drug therapy and achieve therapeutic effects. However, current RMPs focus only on the effects on the fetus and fail to provide suitable protection against adverse events in patients themselves. There is a need to incorporate more information into future RMPs to protect against adverse events in patients. In this study, we evaluated ImrAEs that occur frequently in patients in clinical practice, and their timing of onset. Explaining the symptoms of typical ImrAEs and their timing of onset to patients in RMPs and alerting them can contribute to minimizing the damage caused by ImrAEs. In addition, setting a monitoring period for ImrAEs after the start of IMiD treatment and interviewing of patients by healthcare professionals regarding ImrAEs will lead to early detection of ImrAEs. It is also important in ImrAE management to educate healthcare professionals about typical ImrAEs in the RMP. We hope this study will contribute to improving RMPs in the future.
Limitations
SRSs are passive reporting systems and are, therefore, subject to confounding variables and numerous biases such as under-reporting, over-reporting, and confounders due to comorbidities. Interventions by regulatory authorities may influence the SRS database reporting based on the year of reporting. The influence of differences in medical realities and social backgrounds in each country cannot be ignored. Nomura et al. reported that there were differences in the reported number of AEs between the FAERS and JADER databases; however, the number of shared reports between the FAERS and JADER databases is unknown. 29 The JADER and FAERS databases differ greatly in the number of reports, and results obtained from both cannot be directly compared. Regarding Japanese reports, the JADER database has more reports than the FAERS database, and the JADER database was used to perform the time-to-onset analysis in the Japanese population. Therefore, the JADER database was used for Japan and the FAERS database was used for the US in clarifying ImrAE occurrence in each country. There are considerable differences in the number of reports per country (the US, Great Britain, Germany, France, and Australia) in the FAERS (data not shown). Therefore, the results of this study must be interpreted in light of the differences in the distribution of reporting countries in the FAERS.
In general, adverse events are evaluated based on Common Terminology Criteria for Adverse Events (CTCAE) Grades. 44 However, the SRSs do not provide information on the severity of adverse events. In the future, the ImrAE incidence profile should be investigated taking into account the CTCAE Grade of adverse events.
The FAERS database includes relatively few adverse event reports from Great Britain, Germany, France, and Australia. Therefore, it may not be representative of the ImrAE profile of these countries. Hence, the data of these countries were not evaluated. The results of the analysis using SRSs should be interpreted cautiously based on the assumption that different regulatory rules and medical environments exist in each country.
Conclusions
Despite the inherent limitations associated with SRS data, this study revealed that serious adverse events associated with IMiDs, including myelosuppression-related adverse events, have been reported. By clarifying the ImrAE profile in each country, we obtained information that will lead to the establishment of an appropriate Safety Management Program that focuses on not only preventing fetal exposure but also ensuring the safety of patients. It is desirable to develop an improved version of the RMP that incorporates information on the symptoms of typical ImrAEs and their favorable time of onset, as identified in this study. An improved RMP would contribute to minimizing the damage caused by ImrAEs and improve the quality of life and effectiveness of treatment for patients taking IMiDs.
Supplemental Material
sj-xlsx-1-iji-10.1177_03946320251327618 – Supplemental material for Pharmacovigilance study of immunomodulatory drug-related adverse events using spontaneous reporting system databases
Supplemental material, sj-xlsx-1-iji-10.1177_03946320251327618 for Pharmacovigilance study of immunomodulatory drug-related adverse events using spontaneous reporting system databases by Satoshi Nakao, Mika Maezawa, Moe Yamashita, Koumi Miyasaka, Sakiko Hirofuji, Nanaka Ichihara, Yuka Nokura, Kana Sugishita, Tomofumi Yamazaki, Hirofumi Tamaki, Kimitaka Suetsugu, Masafumi Hashimoto, Toshikazu Tsuji, Kazuhiro Iguchi, Ichiro Ieiri and Mitsuhiro Nakamura in International Journal of Immunopathology and Pharmacology
Supplemental Material
sj-xlsx-2-iji-10.1177_03946320251327618 – Supplemental material for Pharmacovigilance study of immunomodulatory drug-related adverse events using spontaneous reporting system databases
Supplemental material, sj-xlsx-2-iji-10.1177_03946320251327618 for Pharmacovigilance study of immunomodulatory drug-related adverse events using spontaneous reporting system databases by Satoshi Nakao, Mika Maezawa, Moe Yamashita, Koumi Miyasaka, Sakiko Hirofuji, Nanaka Ichihara, Yuka Nokura, Kana Sugishita, Tomofumi Yamazaki, Hirofumi Tamaki, Kimitaka Suetsugu, Masafumi Hashimoto, Toshikazu Tsuji, Kazuhiro Iguchi, Ichiro Ieiri and Mitsuhiro Nakamura in International Journal of Immunopathology and Pharmacology
Footnotes
Acknowledgements
Not applicable.
Authors’ contributions
SN, and MN contributed to the overall concept and design of the study. SN and MN wrote the main manuscript text. SN, MM, MY, KM, SH, NI, YN, KS, and TY conducted data extraction and statistical analysis. HT, KS, MH, TT, KI, and II contributed to the data validation process. MN, KI, and II revised the article critically for important intellectual content. All authors have reviewed the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was partially supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant Numbers 24H02633, 21K11100, and 21K06646). No additional external funding was received for this study.
Ethics approval and consent to participate
Ethical approval was not sought because this was an observational study without any research participants. All results were obtained from data openly available online from the website of the FDA (www.fda.gov) and JADER (![]() ). All data from the FAERS and JADER database were fully anonymized by the regulatory authority before we accessed them. No consent to participate was required owing to the retrospective nature of this study.
). All data from the FAERS and JADER database were fully anonymized by the regulatory authority before we accessed them. No consent to participate was required owing to the retrospective nature of this study.
Consent for publication
Not applicable.
Availability of data and materials
The dataset supporting the conclusions of this article is included within the article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.