
Abstract
Objective
This study investigated the raft-forming suspension of famotidine as an anti-reflux formulation to improve the oral bioavailability of narrow absorption window drugs by enhancing gastric residence time (GRT) and preventing gastro-esophageal reflux disease (GERD).
Method
Various combinations of raft-forming agents, such as Tragacanth gum (TG), guar gum (GG), and xanthan gum (XG), were evaluated alongside sodium alginate (SA) to develop an effective raft. Preformulation studies and preliminary screening were conducted to identify the most suitable raft-forming agent, and GG was chosen due to its mucilaginous properties. The formulation was optimized using a 32 full factorial design, with the quantities of GG and SA as independent factors and apparent viscosity and in-vitro drug release (%) as dependent factors. The in vivo floating behavior study was performed for optimized and stabilized formulation.
Results
Among the tested batches, F6 was selected as the optimized formulation. It exhibited desirable characteristics such as adequate raft weight for extended floating in gastric fluid, improved apparent viscosity, and a significant percentage of drug release at 12 h. A mathematical model was applied to the in-vitro data to gain insights into the drug release mechanism of the formulation. The stability of the suspension was assessed under accelerated conditions, and it demonstrated satisfactory stability. The formulation remains floating in the Rabbit stomach for more than 12 h.
Conclusion
It concludes that the developed formulation has enhanced bioavailability in the combination of GG and SA. The floating layer of the raft prevents acid reflux, and the famotidine is retained for an extended period of time in the gastric region, preventing excess acid secretion. The developed formulations are effective for stomach ulcers and GERD, with the effect of reducing acid secretion by H2 receptor antagonists.
Keywords
Introduction
Gastro-retentive drug delivery systems (GRDDS) have demonstrated advantages over sustained-release formulations. They are particularly beneficial for drugs with a narrow absorption window or those that exhibit high absorption in the stomach but are prone to decomposition in the intestine. 1 GRDDS promotes prolonged retention of pharmaceutical formulations in the stomach, increasing drug solubility for poorly soluble drugs in the intestine, and enhancing absorption and bioavailability. GRDDS allows for higher drug concentrations in the gastric mucosa by releasing drugs locally in the gastrointestinal tract. This could help keep therapeutic drug levels in the systemic circulation. 2
Gastro-esophageal reflux disease (GERD) occurs when gastric contents or bile irritate the esophagus, causing discomfort and the reflux of acidic fluids from the stomach. Weakening or relaxation of the lower esophageal sphincter allows this acid reflux to occur, leading to painful symptoms and potential damage to the esophagus. 3 Smoking, hefty meals, late-night eating, trigger foods, alcohol, coffee, soda, and specific medications taken without consulting a doctor can all aggravate GERD. Symptoms include stomach pain, chest burning pain, dysphagia, persistent laryngitis or hoarseness, throat pain, new-onset asthma, night time coughing, an acidic taste in the throat, regurgitation of food and fluids, and a sensation of a lump in the throat. GERD arises from impaired lower esophageal sphincter function, resulting in acid reflux from the stomach. Currently, available treatments for GERD include antacids, which neutralize stomach acid, medications to reduce acid production such as histamine (H2) blockers (e.g., cimetidine, famotidine, and nizatidine), and proton pump inhibitors like pantoprazole, lansoprazole, and omeprazole. 4 These medications neutralize or prevent acid secretion, but they cannot prevent the reflux of stomach contents into the esophagus. In contrast, the raft-forming drug delivery system prevents the reflux of stomach contents and helps prevent GERD.
Raft development techniques have been effectively employed to prevent its occurrence. 5 Raft-forming drug delivery systems (RFDDS) represent an innovative oral controlled drug delivery approach. When these systems come in contact with gastric fluid, they gel and form a thick, porous layer on the stomach. 6 This relieves acid reflux disorder and slows steady drug release. RFDDS are liquid at ambient temperature but undergo gel formation due to cation-induced gelation and temperature-dependent properties. Raft formation can occur through physical mechanisms involving swelling, diffusion, ion cross-linking, and physiological stimulus based on pH and temperature. This unique mechanism allows the raft formulations to develop a floating layer in the upper part of the stomach, effectively preventing acid reflux while releasing the drug from the delivery system.7–9
Famotidine, classified as a BCS class II drug due to its low aqueous solubility and low intestinal permeability, faces challenges in achieving optimal bioavailability. 10 Only 40%–50% of the drug is absorbed in the upper gastrointestinal tract, with the remaining portion being either poorly available for absorption or completely unavailable. 11 To address this issue, raft-forming drug delivery systems (RFDDS) have been developed to enhance the gastric residence time (GRT) of drugs, thereby improving the bioavailability of famotidine. 12
In RFDDS, sodium alginate is combined with natural mucilages like xanthan gum, guar gum, and tragacanth to form the rafts. Sodium bicarbonate acts as an effervescent mixture, while calcium bicarbonate and carbopol 974 enhance the strength of the rafts. Natural mucilages enhance the GRT of drugs and reduce stomach acidity, which is beneficial for famotidine due to its low bioavailability. Famotidine, an H2 receptor antagonist used for treating ulcers and GERD, exhibits only 40%–50% bioavailability. 13 Combining famotidine with antacids promotes absorption by the receptors in the parietal cell wall. GRDDS, employing rafting technology, helps retain the drug in the stomach for an extended period, enhancing its bioavailability.
Materials and methods
Famotidine obtained as gift sample from Steril Gene Life Sciences (P) Limited. All the other required excipients like sodium alginate (SA), xanthan gum (XG), guar gum (GG), carbopol, and tragacanth were purchased from Sigma Aldrich, Chennai.
Preformulation studies
Solubility study
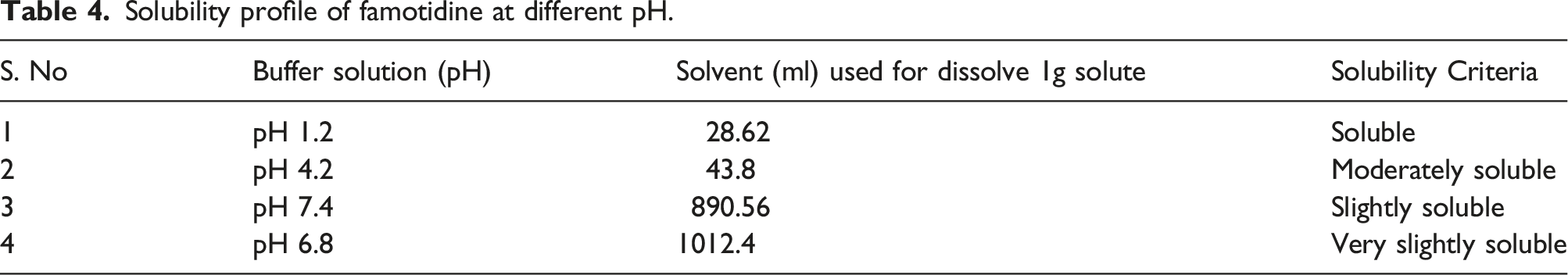
To determine the solubility profile of famotidine, the shake flask method was employed. Initially, 100 mL of a solubilizing medium was transferred into a 250-ml conical flask, followed by the addition of an excess amount of famotidine. The experiment was conducted at a controlled temperature of 25°C. The conical flask, containing the solubilizing medium and famotidine, was placed on a rotary shaker operating at 200 RPM for a period of 24 h.
After the incubation period, a portion of the famotidine solution was solubilized in buffer solution. Using a UV-visible spectrophotometer, the absorbance of the solubilized famotidine was measured at a specific wavelength of 265 nm. This measurement allowed for the estimation of the quantity of famotidine dissolved in the solubilizing medium, which was then recorded. 14
To ensure accuracy and reliability, the solubility study was repeated three times using different buffer solutions with varying pH values, including pH 1.2, 4.2, 6.8, and 7.4. Each repetition allowed for the assessment of famotidine’s solubility under distinct pH conditions.
Calibration curve for famotidine
Solution I: In a 100-ml volumetric flask, 100 mg of famotidine was solubilized with the required amount of 0.1 N HCl. The remaining volume was then filled with 0.1 N HCl, resulting in a concentration of 1 mg/mL or 1000 µg/ml.
Solution II: From Solution I, 10 mL was withdrawn and diluted with 0.1 N HCl in a 100-ml volumetric flask, producing a concentration of 100 µg/ml. Subsequently, aliquots of 0.2, 0.4, 0.6, 0.8, and 1 mL were withdrawn from this diluted solution and further diluted in 10 mL volumetric flasks with 0.1 N HCl. This resulted in concentrations of 2, 4, 6, 8, and 10 µg/ml, respectively.
Using a UV-visible double beam spectrophotometer, the absorbance of each diluted solution was measured at a wavelength of 265 nm. 15 This data was then utilized to construct the calibration curve for famotidine, allowing for accurate determination of its concentration in subsequent samples based on their absorbance values.
Drug and excipients compatibility studies
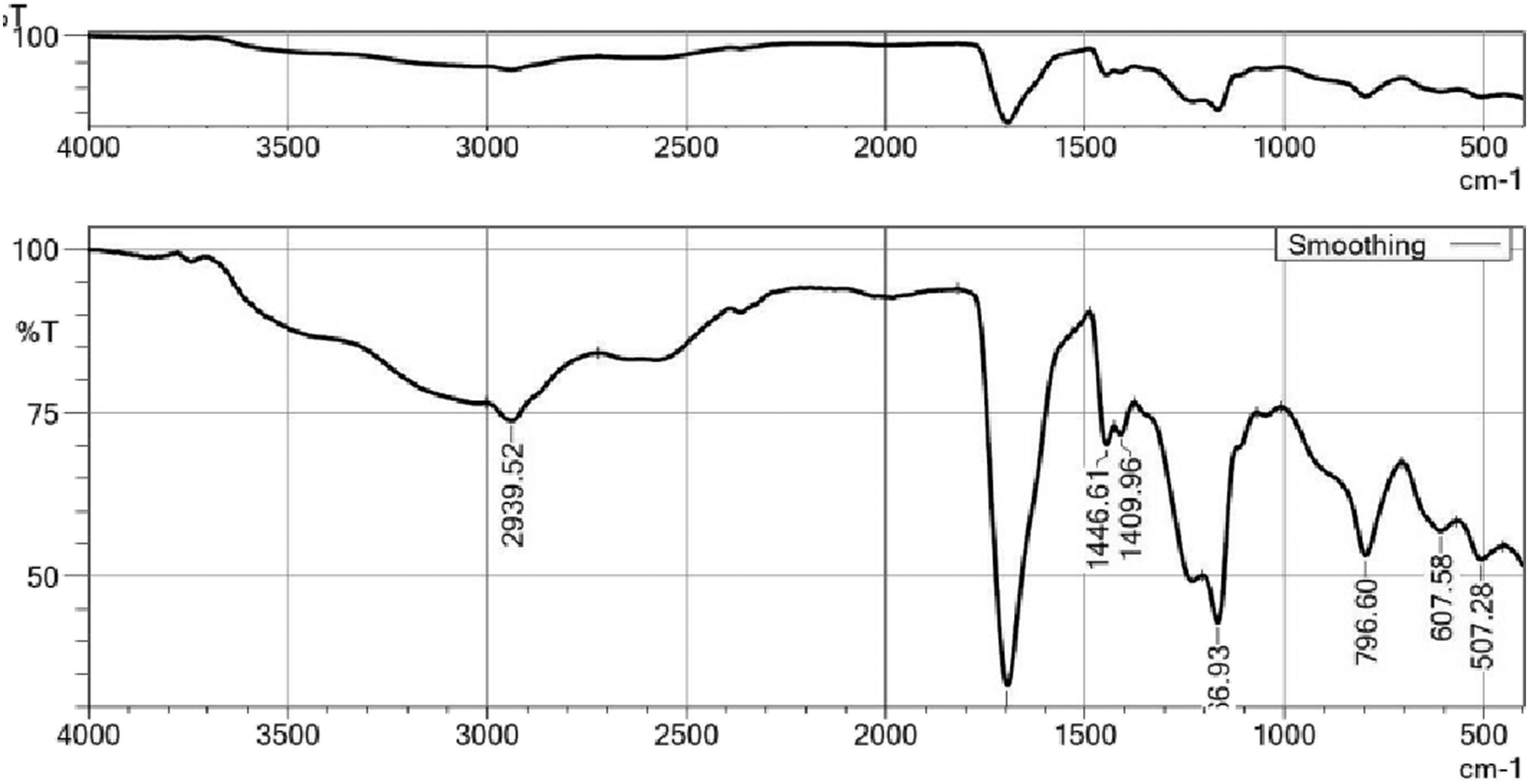
Drug-excipient compatibility was determined by analyzing the interaction between the FTIR spectra of famotidine and the FTIR spectra of the drug with excipients. Compatibility study by FTIR Shimadzu IR-Tracer 100 performed with the KBr pellet method. The testing samples were uniformly mixed with KBr crystals, and then they were compressed as a disc. The spectrum was taken by placing the disc on the FTIR spectrophotometer. 16
Method of preparation
Formulation of famotidine suspension by RFDDS

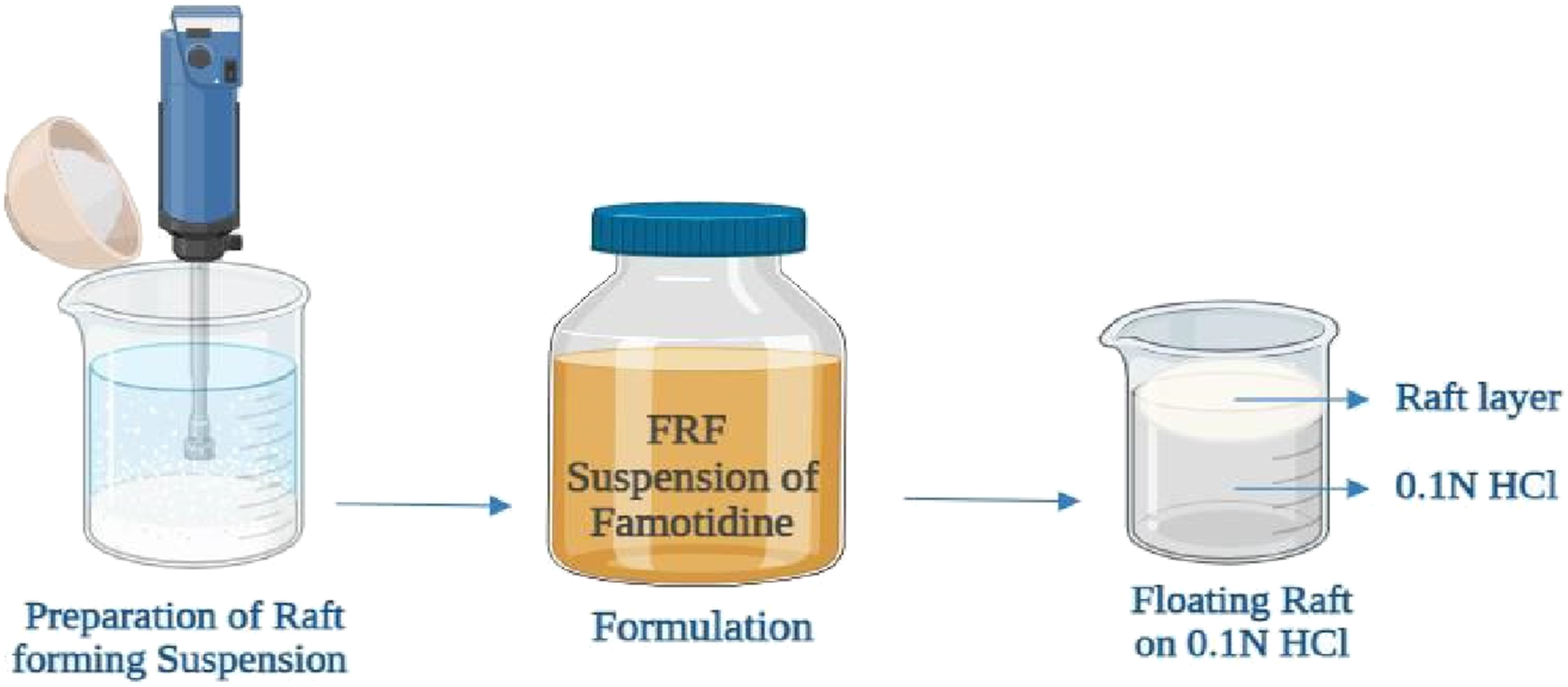
Process of raft development of famotidine suspension.
Preliminary studies
To enhance the raft weight, in-vitro gelation time, in-vitro dissolution studies, and ANC (acid-neutralizing capacity), preliminary studies were conducted. The aim was to select an appropriate natural gum, such as tragacanth, guar gum, and xanthan gum, in combination with sodium alginate. Eight batches were prepared and evaluated to determine their performance.
After analyzing the results of the preliminary studies, it was determined that the combination of guar gum and sodium alginate showed promising outcomes. Hence, this combination was chosen for further optimization using 32 full factorial designs. The goal of the optimization process was to enhance the desired characteristics of the raft-forming drug delivery system.
Optimization of a selected batch
Coding of variable for 32 full factorial design.

Optimization by three-level two-factorial design (batch G2).

Evaluation of famotidine suspension
In-vitro gelation time
10 mL of formulation was added to a 250-ml beaker, which contains 100 mL of 0.1 N HCl, to determine in vitro gelation time. The time needed to form a raft on the solution and clear the lower part of the beaker is known as in-vitro gelation time. 18
Floating time
10 mL of formulation was added to a 250-ml beaker, which contains 100 mL of 0.1 N HCl, to determine the floating time. This is described by the extensive time period during which the raft remains floating on the liquid. 19
Raft weight
For this test, 100 mL of 0.1 N HCl was taken in a 250 mL beaker. In this, 10 mL of formulation was poured; raft lefts developed uniformly. After complete development of the raft, the beaker is kept to one side for 30 min, and any leftover liquid in the beaker is decanted carefully. The raft was collected from the beaker and transferred to the butter paper; excess liquid in the raft was removed by using a paper towel, and the raft dried for 2 h. Afterward, the dried raft weight was calculated and noted. 20
Raft volume
The empty beaker was weighed after it had dried completely and written as W1. 100 mL of 0.1 N HCl (raft-developing liquid) was transferred into a beaker, marked for the level of liquid, then weighed and written as W2. In this beaker, 10 mL of formulation was added. Then allow it to develop into a raft and keep it aside for a few minutes. Afterward, leftover liquid was drained, and the raft was collected in a butter paper bag and dried with paper towels. Then, the raft was weighed and written as W3. Purified water was filled up to the mark made in the same beaker used for raft development while 100 mL of raft-developing liquid was added; in this dried raft, weight was taken and written as W4; with all the collected data, raft volume was estimated by the below formula
21

Raft strength
Sufficient strength is necessary to prevent the reflux of the stomach contents into the esophageal region. To assess this, 10 mL of the formulation was transferred into a 250 mL glass beaker containing 150 mL of pH 1.2 buffer. The raft was allowed to develop and kept aside for 30-min with an L-shaped wire probe which was immersed straight within the beaker. The strength of the resulting raft was determined using the modified balance method. Water was added drop by drop to the pan, and the amount of water needed to break the raft was recorded. 22
Raft resilience
The raft was formed in a 250 mL glass jar by adding a bilayer tablet to 150 mL of SGF pH 1·2 previously maintained at 37°C. The raft was allowed to develop for 30 min; the capped jar was positioned in a modified cube mixer, set to revolve at 20 rpm, to simulate gastric agitation. The raft was assessed visually for such a time that a raft could no longer be detected. A raft was distinct or dispersed into two or more hovering gels at least 15 mm dia. 23
Acid neutralization capacity (ANC)
A tablet powder equivalent to unit dose was taken in a 250-ml beaker. Water was added to make a total volume of about 70 mL, heated to 37°C and stirred continuously by maintaining the temperature at 37°C. 30 mL of 1M hydrochloric acid (previously heated to 37°C) was added and mixture was maintained at 37°C for 15 min with continuous stirring. The excess acid was titrated with 1M sodium hydroxide to a pH of 3.5. The number of meq of acid consumed by the tablet tested was calculated by the following formula 24 :
Total mEq = (V HCl *N HCl) – (V NaOH *N NaOH)
Where, M HCl = molarity of hydrochloric acid
M NaOH = molarity of sodium hydroxide
V NaOH = volume of sodium hydroxide
Dissolution studies
The in-vitro drug release data was determined by the USP Type II dissolution apparatus. The paddle shaft was rotated at 25 rpm, and the chamber was maintained at 37°C. For this study, 900 mL of 0.1 N HCl was taken in a basket. 10 mL of the formulation was placed in the dissolution testing basket, and the raft was left to develop uniformly. 11 Then the paddle began to rotate at 25 rpm, and from the basket, 1 mL of solution was collected in 10 mL standard flasks at every 30 min interval, and the volume was filled up to the level with 0.1 N HCl. Replace the volume with 1 mL of 0.1 N HCl. Using a UV-visible double-beam spectrophotometer, the absorbance was analyzed at 265 nm to get the concentration by applying a calibration factor.25,26
In-vitro diffusion study
A Keshary–Chien-type diffusion cell was used for the release study. The cellophane membrane, or dialysis membrane, was immersed in phosphate buffer overnight. Later, the membrane was mounted between the donor and receptor compartments. Micro-bead placed inside the receptor compartment to maintain the hydrodynamics of the fluid at 400 rpm. The compartments were clamped together, and the temperature was maintained at 37°C. Receptor compartment filled with pH 7.4 phosphate buffer. 1 mL of samples was withdrawn at particular time intervals. The same volume was replaced with buffers to maintain sink conditions. The absorbance of samples was analyzed in a UV-visible spectrophotometer.27–29
In vivo floating behavior by X-ray imaging
The study was performed after obtaining approval from the Institutional Animal Ethics Committee (IAEC). The in vivo floating behavior of floating raft formulation was analyzed by X-ray imaging technique were performed in Albino rabbits. Barium sulphate-loaded floating raft formulation was administered under fasting, and compared with marketed formulation. The X-ray images of rabbits can indicate whether a selected formulation is capable of developing a raft in the stomach. Through these images the in vivo floating behaviour of guar gum can be determined.30–32
Results and discussion
Preformulation studies
Solubility profile of famotidine at different pH.
Statistical analysis of the calibration curve.

The profiles of the drug and excipients provided valuable insights for the formulation development. Compatibility testing between the drug and excipients involved evaluating their physical characteristics and potential chemical reactions. The FT-IR analysis revealed no significant interaction between the drugs and excipients, as depicted in Figures 2 and 3. This finding confirmed that the selected excipients were suitable for the development of the raft-forming suspension of famotidine. FTIR spectra of famotidine. FT-IR spectra of formulation.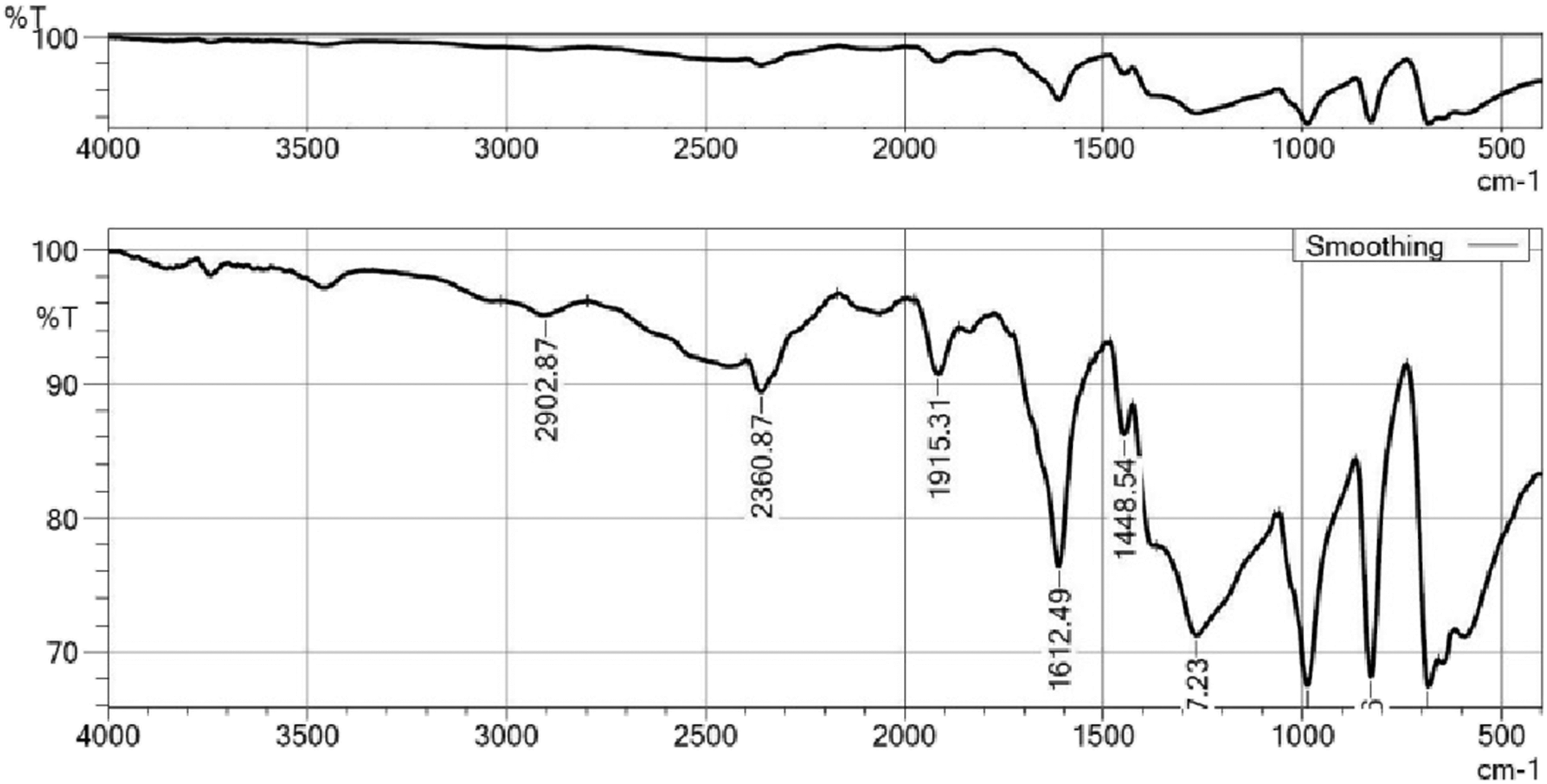
Surface morphology
The particle size of the optimized raft-forming suspension of famotidine is around 1 to 100 μm (Figure 4). The morphology of the raft-forming suspension was analyzed by SEM (Figure 5). The photograph was examined at higher resolution for the optimized formulation through a scanning electron microscope. Particle size analysis. SEM image of formulation (F6).

Results of preliminary batches
Comparative results of preliminary studies in raft-forming suspension of famotidine.

n = 3*.
Results of optimized batch
Comparative results of optimized batch in raft forming suspension of famotidine.

n = 3*.
Among the optimized formulations, F6 demonstrated better in-vitro drug release (91.40 ± 0.12) at 12 h and a higher ANC (7.44 ± 0.3) compared to other formulations. Thus, F6 showed a more significant impact on raft development, exhibiting suitable pH (8.41 ± 0.1), in-vitro gelation time, raft volume (3.29 ± 0.22), viscosity (1480 ± 3), ANC, and in-vitro drug release. The comparative study of percentage drug release for all optimized batches is illustrated in Figure 6. Additionally, the optimization results of the raft-forming suspension using sodium alginate and guar gum, including contour plots and response surface plots of ANC and in-vitro drug release (%), are depicted in Figure 7 (a)–(d). Model statistics for responses Y1 and Y2 (raft-forming suspension using sodium alginate and guar gum) are presented in Table 8. In-vitro percentage drug release of optimized batch. (a), (b), (c), and (d): Contour plot and response surface plot of ANC and in-vitro drug release (%) in raft-forming suspension using sodium alginate and guar gum. Model statistics for responses Y1 and Y2 (Raft-forming suspension using sodium alginate and guar gum).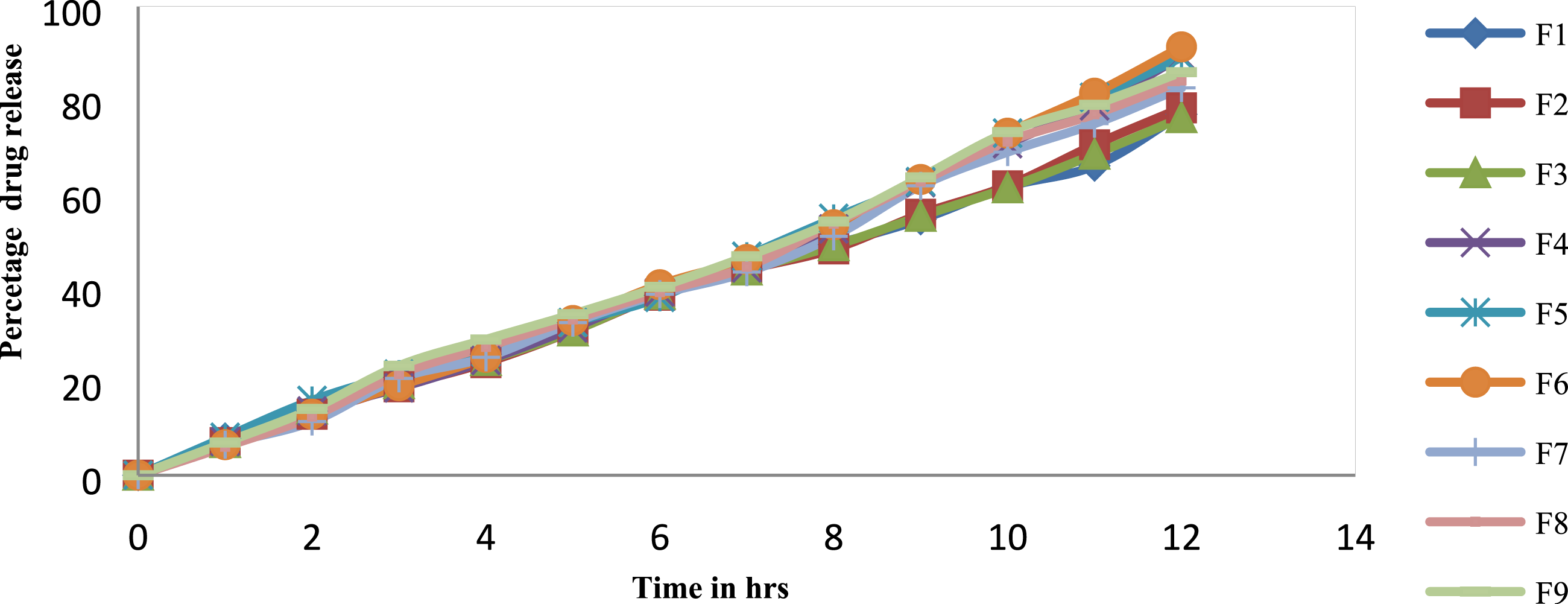


The regression equations for the raft-forming suspension using sodium alginate and guar gum were determined using the response surface method.
Y1 = 22.29-0.6250*X1-9.25*X2
Y2 = 82.08 + 1.76*X1-0.9275*X2+2.08*X1*X2*-0.4932*X12 -0.3882*X22
Mathematical model of optimized batch F6.


Kinetic model for optimized batch (F6) of raft-forming suspension of (a) Zero order, b) First order, (c) Korsmeyer peppas and (d) Higuchi plot.
In-vitro drug diffusion studies
In-Vitro diffusion studies of pure drug and batch F6.


In-vitro diffusion study of pure drug and formulation (Batch F6).
The raft strength was ranged from 6·390 ± 0·067 g to 18·590 ± 0·057 g. The raft thickness ranged from 2·50 ± 0·11 to 4·50 ± 0·36 cm. The F6 formulation showed a greatest raft thickness due to the maximum amount of SA-GG. The thickness of the raft increased as the concentration of the polymers increased. A direct relationship between the raft thickness and strength was observed. When the raft thickness was increased, the strength of the raft increased.
Stability studies
Stability studies for optimized batch F6.

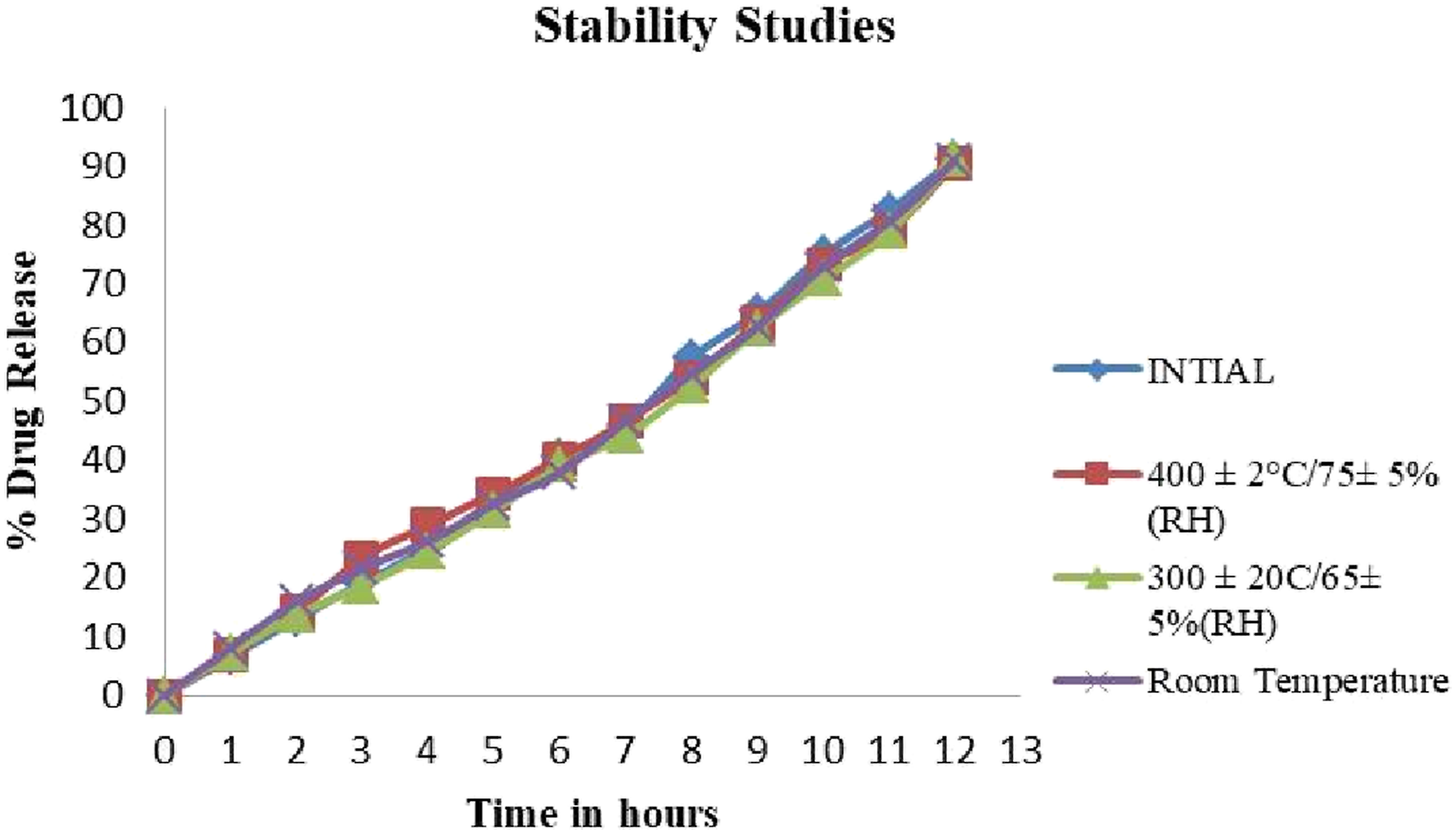
Comparative study of In-Vitro drug release time before and after stability.
In vivo floating behavior by X-ray imaging study
The X-ray images were taken after the rabbits were administered a barium sulphate-loaded, optimized raft-forming formulation during fasting-state and fed state. Figure 11(a)–(c) depicts X-ray images of the rabbit for empty stomach, 0 h, and 12 h respectively. The in vivo floating behavior of raft-forming suspension retained in the stomach for more than 12 h. This confirms that factors include fed and unfed state not affecting the formulation’s floatability in stomach and the drug well entrapped in the formulation, which releases the drug over 12 h. Other important factor adhesive index of the polymers (guar gum) in the formulation is high, this creates a layer on stomach wall explained by Rahamathulla M et al, 2021,
33
and famotidine does not have the adhesive properties to adhere to the stomach mucosa. However, the polymer and other elements in the formulation are entrapped during raft development; the adhesive index has no impact on the formulation’s gastric residence time. Famotidine possesses a limited half-life and is readily absorbed from the stomach mucosa, yet it exhibits a low gastric residence time and it does not have the adhesive properties to adhere to the stomach mucosa. The findings have substantiated that the formulation of a raft-forming suspension of famotidine enhances the gastric residence time (GRT) and ensures the availability of the drug for effective absorption. (a) Empty stomach (b) Initial raft development (0 h) (c) At 12 h.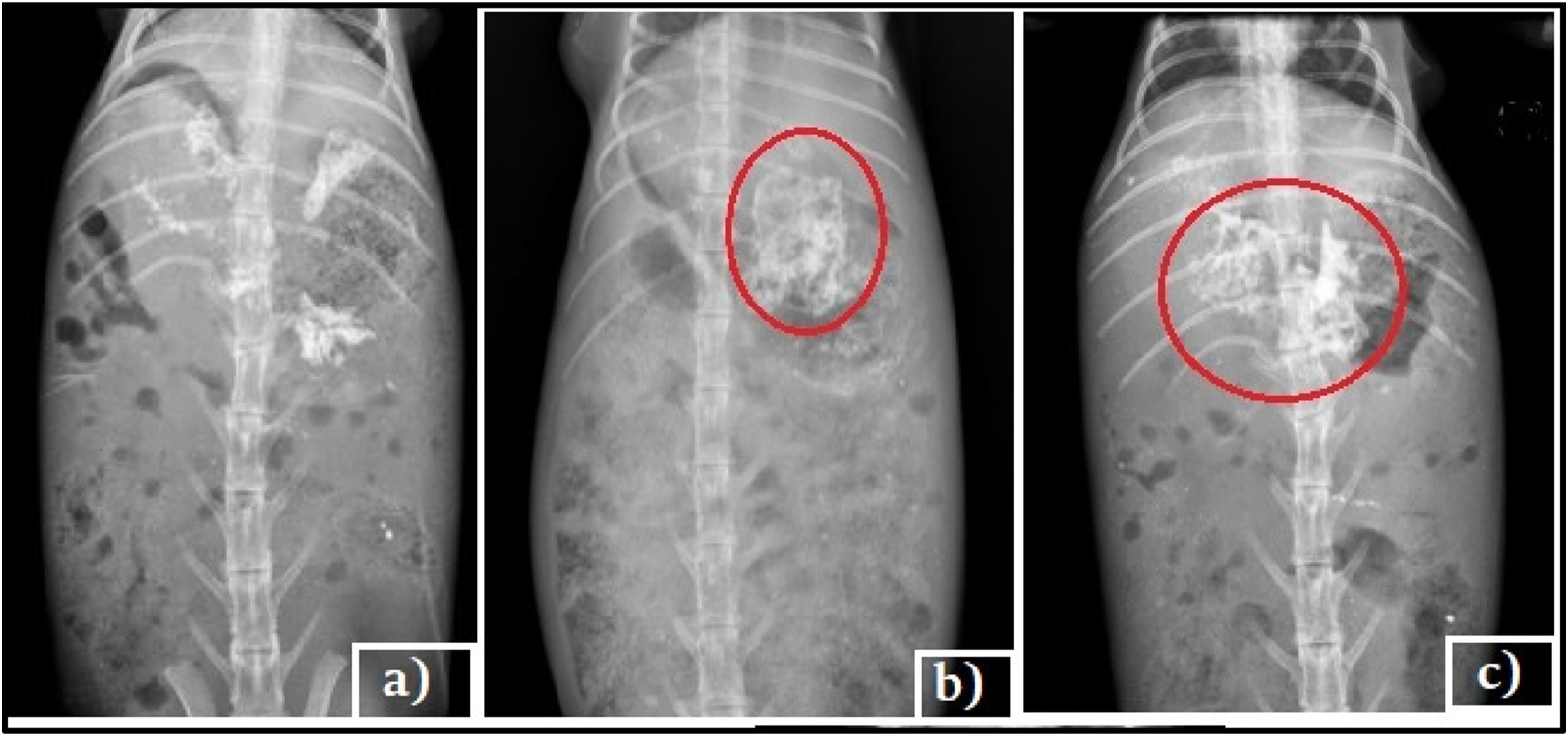
Conclusion
In conclusion, a famotidine suspension formulated using a raft-forming drug delivery system with guar gum and sodium alginate as the raft-developing polymers has been successfully developed. The inclusion of calcium carbonate and sodium bicarbonate as acid-neutralizing agents further enhances the formulation. When the suspension comes into contact with 0.1 N HCl, the release of CO2 triggers the formation of a floating raft within a desirable floating lag time of 21 ± 0.3 to 45 ± 0.5 s. Optimization of the formulation was achieved using a three-level, two-factorial design, with the combination of guar gum and sodium alginate (GG-SA) demonstrating improved floating lag time, pH, raft volume, raft weight, and acid neutralization capacity. The optimized formulation was able to release the drug effectively for a sustained period of 12 h. Kinetic analysis revealed that the famotidine release from the raft-forming drug delivery system follows zero-order kinetics, indicating controlled and prolonged drug release. The FT-IR analysis confirmed no significant interaction between the drugs and excipients, ensuring formulation stability. The optimized formulation remained stable under accelerated conditions of temperature and humidity. The controlled release of famotidine from the GG-SA formulation exhibited a significant effect on the absorption window, leading to improved bioavailability by extending the gastric retention time of the drug in the stomach. The in-vitro drug diffusion results further support the enhanced drug release achieved with the F6 formulation. The in vivo floating behavior study confirms the floatability of raft in the stomach for more than 12 h. Overall, the development of a raft-forming suspension using GG-SA provides a promising approach for enhancing bioavailability and optimizing the treatment of gastro-esophageal reflux disease (GERD) and stomach ulcers.
Footnotes
Acknowledgements
The authors would like to thank Department of Pharmaceutics, SRM College of Pharmacy, SRMIST, Kattankulathur, for providing facilities and guidance to carry out the research.
Author contributions
Material preparation, data collection, and analysis were performed by [Rajalakshmi Munusamy], and [Sangeetha Shanmugasundharam]. The first draft of the manuscript was written by [Rajalakshmi Munusamy] and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.