
Abstract
Malignant tumors of the female reproductive system are a serious health and social problem, as they are the second most common cause of death among women, after breast cancer. Their incidence has increased dramatically during recent years, probably due to the different sexual habits and changes in the prevalence of HIV/ AIDS and HPV virus carriers, among other factors. Vulvar tumors represent only 4% of all gynecological neoplasms, and they are fourth in frequency after tumors of the cervix, uterus, and ovary. Ninety eight percent of all vulvar tumors are benign and only 2% are malignant. The overall incidence of tumors with vulvar location is between two and seven cases per 100,000 women, and it increases with age, while the death rate is estimated at 0.7 per 100,000 women. Sarcomas of the vulva comprise approximately 1–3% of all vulvar cancers, with leiomyosarcomas, epithelioid sarcomas, and rhabdomyosarcomas being the most common among them. They are characterized by rapid growth, high metastatic potential, frequent recurrences, aggressive behavior, and high mortality rate. In this paper, we present the most common forms of sarcomas of the vulva (leiomyosarcoma, epithelioid sarcoma, malignant rhabdoid tumor, rhabdomyosarcoma) in order to emphasize the broad differential diagnosis, rare appearance, non-specific clinical picture, aggressive course, and high mortality.
Introduction
Sarcomas are comparatively rare tumors, accounting for about 2% of all cancers in human pathology.1,2 They are neoplasms with mesenchymal origin and may arise from the soft tissues and viscera. Soft-tissue sarcomas comprise less than 3% of all tumors in the female reproductive system. 1
Uterine sarcomas represent 90% of all gynecological sarcomas, while sarcomas with vulvar localization account for only 1–3% of all vulvar cancers.
The most common vulvar sarcomas are leiomyosarcomas, rhabdomyosarcomas, angiosarcomas, malignant peripheral nerve sheath tumors, and ‘malignant fibrous histiocytomas’; aggressive angiomyxoma is sometimes included in this group.1–3
Vulvar sarcomas are characterized by non-specific clinical manifestations often leading to misdiagnosis, most commonly as Bartholin’s cysts or abscesses. 3 The median age of occurrence is 50 years, but the interval is wide, depending on the morphological variant. Usually they are asymptomatic or present with non-specific local discomfort with vulvar mass, or chronic vulvar pruritus of longstanding, while pain, bleeding, or ulceration are late symptoms associated with poor prognosis. 3
The prognosis depends mainly on the size of the tumor, involvement of adjacent tissues, and mitotic activity. It is generally poor, as vulvar sarcomas are typically characterized by rapid growth, high metastatic potential, frequent recurrences, aggressive behavior, and high mortality rate.1–3
Leiomyosarcoma of the vulva
Leiomyosarcoma of the vulva (LMS) is the most common histologic variant of sarcoma with vulvar localization, accounting for approximately 1% of all tumors in this area.1,2 Morphologically, leiomyosarcoma may arise from smooth muscles, blood vessels, rough ligaments, and erector-pili muscles.1,3 The tumor usually arises in the area of Bartholin’s gland, at the usual place of Bartholin duct cysts, and shows clinical similarities with them, leading to difficulties in diagnosis. 4
LMS occurs more frequently in women of middle and older ages (age range, 20–68); the average age at clinical presentation is approximately 30–40 years, and the youngest patient described was aged 14 years.5,6 The majority of these cases are from Western countries, which suggests etiopathogenic connection with lifestyle or a certain genetic predisposition. 5 The role of chronic inflammation as a precursor of carcinogenesis is debated. Indeed cases associated with lichen sclerosus have been reported. 7 Manifestation of LMS during pregnancy is possible, and for early diagnosis and timely therapy histological confirmation of tumors is still needed during pregnancy. 8
It clinically presents as a solid, infiltrating tumor mass, with diameters in the range of 1.5–16 cm (average size, 5 cm). 4 Subjectively the lesions are usually painless and asymptomatic, but in advanced stages they can be associated with voiding dysfunction or bleeding. 2
LMS shows immunoreactivity to muscle markers, which is also typical for leiomyomas; 4 namely, with desmin, smooth muscle actin, and muscle-specific actin. Focal positivity for S-100 and cytokeratin is possible.1,4 Immunohistochemical expression of estrogen, progesterone, and androgen receptors, in addition to a moderate number of Ki-67-positive cells and absence of p53 protein overexpression, are markers of low histologic grade of vulvar leiomyosarcoma.1,9 thus not requiring the use of adjuvant radiotherapy. 9
The diagnostic criteria that come into consideration in the differentiation of extrauterine leiomyosarcomas from leiomyomas are: (1) diameter >5 cm; (2) infiltrative margins; (3) >5 mitotic figures per 10 HPF; and (4) moderate to severe cytologic atypia (Figures 1 and 2). The presence of at least three of the above-mentioned criteria supports the diagnosis of LMS.9,10 According to data from a 30-year retrospective study, mitotic activity is defined as an important prognostic marker associated with an increased risk of relapse. 11

Well-differentiated leiomyosarcoma. Fascicles of spindle-shaped cells with eosinophilic cytoplasm and cigar-shaped nuclei; focally hyperchromatic cells can be seen (hematoxylin/eosin, original magnification x100).
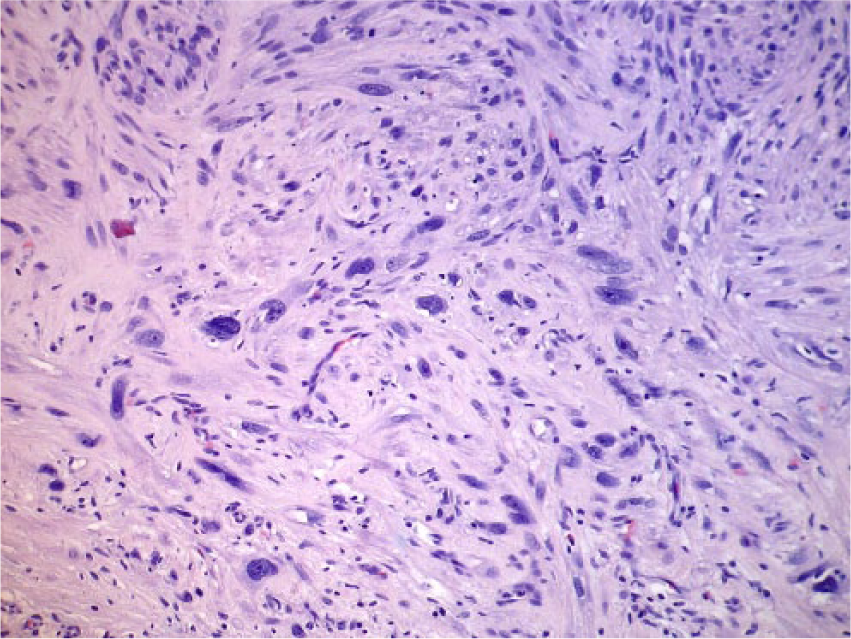
Well-differentiated leiomyosarcoma. Pleomorphic cells, displaying nuclear hyperchromasia, and frequent nucleolar prominence (hematoxylin/eosin, original magnification x200).
The recommended therapeutic approach is complete surgical excision (i.e. negative margins confirmed by pathology), followed by radiation therapy, although there are no unified algorithms for the treatment of this type of sarcoma.12,13 The administration of chemotherapy is not considered mandatory, but some evidence suggests that it may reduce the chances of relapse and the risk of developing metastatic disease. 14 Without the presence of metastasis, the prognosis for a completely excised tumor is good.13,14
Myxoid leiomyosarcoma of the vulva (MLMS) is an extremely rare mesenchymal tumor, with fewer than 10 cases described in the literature. 15 MLMS is typical of elderly women, but there have been reported cases of occurrence during pregnancy. 16 It presents clinically as a unilateral non-ulcerated, slow-growing, painless vulvar mass. 15 It is possible that an initial biopsy might not be representative, showing histological findings suggestive of a benign tumor.15,17 This tumor requires radical surgical excision with subsequent histological examination of the entire lesion. 16 Two cases of myxoid leiomyosarcoma of the Bartholin gland have been reported. 17
Epithelioid sarcoma (ES) is a very aggressive undifferentiated soft-tissue sarcoma with extremely rare presentation on the vulva (2% of cases).18,19 About 20 cases in the world literature have been described; more than half of them have a poor prognosis, while single cases were reported as successfully treated. 18
‘Proximal’ type epithelioid sarcoma (PES) and malignant rhabdoid tumor (MRT) are believed to represent closely related lesions, probably belonging to the same clinicopathologic spectrum. 18 Recent studies indicate that immunoreactivity for CA125 and loss of INI-1 expression are useful immunohistochemical features in the diagnosis of epithelioid sarcomas. 20 The prognosis is poor, with survival rates of 73%, 55%, 27%, and 9% at 1, 2, 5, and 10 years, respectively.19,20 The median survival is 27 months after diagnosis. 21 Due to the rare appearance of this type of neoplasm in this location, there are no unified data and algorithms regarding therapeutic approaches. 22 Early diagnosis and radical surgical resection, followed by adjuvant therapy, have been reported as an optimal therapeutic option.18,22
The conventional, ‘distal-type’ epithelioid sarcoma is usually localized in the labia majora, and it is more common in women of reproductive age. 23 The age varies over a wide range, 23–80 years, averaging 36 years. 24 Individual cases have been described as having manifested during pregnancy. 23
The molecular pathogenesis of this type of neoplasm remains unclear. 19 It usually occurs clinically as an asymptomatic nodular mass with dimensions in the range of of 1–10 cm (on average, about 5 cm in diameter), resulting in a delay in diagnosis, which increases the risk of invasion of neighboring structures. 25 A granuloma annulare-like pattern is a characteristic histological finding, with a moderate or high level of cellular atypia. 22
‘Distal-type’ epithelioid sarcoma has high metastatic potential and metastasizes early, most commonly to the lungs, frequently leading to organ failure.23,24 Recurrences are common and the mortality is high.25,26 Immunohistochemical study is appropriate to confirm the diagnosis, and includes immunoreactivity for vimentin, cytokeratin (CK), and epithelial membrane antigen (EMA) in most cases. 27 Unusual SMARCB1/INI1 gene abnormalities in epithelioid sarcoma have been also detected, which can be helpful in differentiation from malignant rhabdoid tumor. 25
Early diagnosis and prompt treatment are essential for improvement in prognosis. 18 ES, diagnosed in early stages of tumor invasion, shows a favorable prognosis and median survival averaging 21 months, while in stages II and III, it is only 6 months. 28
Definitive excision with a margin of at least 2 cm, hemivulvectomy with bilateral inguinal lymph node dissection, followed by adjuvant chemoradiotherapy, and chemotherapy are the possible treatment options.21,22 Radical vulvectomy with flap reconstruction is described to have greater efficacy in terms of survival. 23 The prognosis is poor because of the aggressive nature of the tumor. 29 According to a survey, 86% of patients relapse, 29% develop early lymph node metastases, and 64% die within 3 years after diagnosis. 21
Proximal-type epithelioid sarcoma represents a rare variant of this type of sarcoma with features of both the ‘atypical’ epithelioid sarcoma, and a rhabdoid tumor or undifferentiated carcinoma. 21 It is characterized by an aggressive course and high mortality.30,31
It presents clinically with a rapidly growing, asymptomatic tumoral mass, with a high degree of infiltrative growth; a multinodular pattern is apparent in some cases.32,33 The size of the lesions is in the range of 1–20 cm in diameter, with an average of about 4 cm. 32
Histological examination shows epithelioid, rhabdoid cells with vesicular nuclei, large prominent nucleoli, and cytoplasmic eosinophilic globules, separated by thin, fibrous septa and areas of necrosis.34,35 They are characterized immunohistochemically by diffuse positivity for vimentin and EMA, and focal positivity for cytokeratin. 36 They are usually negative for CK5/6, CD34, S-100 protein, desmin, and myogenin. 36 This type of sarcoma shows inactivation of INI1 (hSNF5/SMARCB1, a member of the SW1/SNF chromatin remodeling complex located on chromosome 22q11.2) – a finding that is typical of rhabdoid tumors of infancy. 18 Ultrastructurally, some cases show a rhabdoid phenotype, with prominent intracytoplasmic intermediate filament aggregates, often accumulating into paranuclear whorls, while others are characterized by features of epithelial differentiation with tonofilament-like structures or desmosomes; still other cases show a mixture of both phenotypes.18,20
Conventional epithelioid sarcoma, extrarenal malignant rhabdoid tumor, epithelioid malignant peripheral nerve sheath tumor, melanoma, rhabdomyosarcoma, and undifferentiated carcinoma should be considered in the differential diagnosis.30,31
The prognosis is poor, as four of five patients die due to multiple organ failure resulting from distant metastases. 32 Multiple recurrences and early metastases are typical for this type of tumor, as the majority of patients have distant metastases at the time of diagnosis of the recurrences. 35 Median survival is on average 19 months. 18 Unlike the conventional, ‘distal-type’ epithelioid sarcoma, the proximal variant is characterized with dominant large-cell, epithelioid cytomorphology, striking cytologic atypia, frequent occurrence of rhabdoid features, and lack of a granuloma-like pattern.21,34 It has a significantly poorer prognosis, metastasizes faster, and results in higher and premature mortality.31,34
Malignant rhabdoid tumor of the vulva
Malignant rhabdoid tumor of the vulva (MRT) is a highly aggressive tumor that is rarely localized outside the kidneys and central nervous system.37,38 Vulvar location is extremely rare with fewer than 15 cases reported. 37 Described for the first time in 1978 by Beckwith et al. as a rare rhabdomyosarcomatous subtype of Wilms’ tumor, 39 MRT has been recognized as a separate variant only since 1981. 40
MRT is a tumor typical of childhood, and the pathogenesis is unclear. 18 Vulvar location, however, is more typical for the age group in the range of 30–50 years. 41 It is usually observed in the labia majora, mons pubis, and clitoris. 37 It clinically presents as an asymptomatic, well-circumscribed, solid tumor mass, with size in the range of 2–10 cm. 39
The diagnosis is confirmed histologically by the characteristic finding of diffuse proliferation of ‘rhabdoid cells’; that is, rounded or polygonal cells with eccentric nuclei, prominent nucleoli, and glassy eosinophilic cytoplasm containing hyaline-like inclusion bodies, arranged in sheets and nests. 41 These characteristic rhabdoid cells may be present in other soft tissue tumors, such as synovial sarcoma, extraskeletal myxoid chondrosarcoma, and leiomyosarcoma, and their existence is a marker of poor prognosis. 39 Most MRTs express vimentin, followed by EMA, cytokeratin, and CD 99, but not desmin. 37
The differentiation of the MRT from epitheloid sarcoma is difficult, sometimes impossible, because of their overlapping clinical and histological features. 42 According to some authors, MRT is a variant of epithelioid sarcoma, with a more aggressive course and a poor prognosis. 41 MRT shows immunoreactivity for CK8 and CK18, while classical type ESs are diffusely positive both for CK8 and CK18, and other low molecular weight cytokeratins such as CK4, 6, 10, 13, 16, 17, as well as ‘high-molecular-weight’ CKs such as CK1, 5, 10, and 14. 43 Abnormalities in the long arm of chromosome 22 have been identified by cytogenetic analysis with alteration of the hSNF5/INI1 (SMARCB1) gene in renal, extrarenal, and intracranial MRT. 37 These variations, however, have also been seen in proximal-type epithelioid sarcoma. 37 Loss of SMARCB1/INI1 protein expression is considered to be a marker for histological confirmation of the diagnosis of MRT. 41 It is found, however, in ES, although at a lower frequency, and different mechanisms are considered to lead to these abnormalities in the two sarcomas. 41 INI1 antibody immunohistochemistry has also proved useful in the confirmation of the histological diagnosis of MRT.40,42
Therapy consists of surgical excision with or without lymph node dissection.42,43 Relapses are common, with an average latency of several years after diagnosis. 38 Excision followed by radiotherapy is the recommended therapeutic approach in these cases of recurrence. 39 The development of distant metastasis is also frequent and, in most cases, proves to be fatal. 37 Radical surgical excision and the use of actinomycin have been reported with good efficacy. 44 There is no proof that radiotherapy or chemotherapy influences survival. 45 The prognosis is poor, mortality is high, and the median survival is 9 months after diagnosis. 37 According to survey data, advanced age is typically associated with a more favorable prognosis. 46
Rhabdomyosarcoma of the vulva
Rhabdomyosarcoma (RMS) is the most common type of soft tissue sarcoma in childhood, mainly under the age of 15 years. 47 It arises from skeletal muscle and can affect any part of the body where this type of muscle can occur.47,48 During childhood, a genital location for this tumor is second most common, following the head and neck, and occurs primarily on the vagina or uterus, rather more rarely on the vulva; it is extremely rare in adults.49,50
First described by Weber in 1854, RMS was recognized as a separate entity 92 years later by Stout, in 1946, as a tumor with an aggressive course of unknown cause.48,50
Early onset is typical of RMS, frequently before the age of 5 years, with aggressive behavior and predominantly affecting the male gender. 51 Forty percent of the cases are located in the head and neck, and only 25% of all RMS are located on the genitalia – vulva, vagina, bladder, or cervix.48,51 Although vulvar location is rare, it is associated with a more favorable prognosis.48,52
Most of the cases occur sporadically, but genetic factors have been found in about 30% of patients, and they appear to be a risk factor for the development of RMS. 47 These include the presence of the rare familial syndromes such as Li-Fraumeni syndrome, which includes familial clustering of RMS and other soft tissue tumors in children, as well as adrenocortical carcinoma and early-onset breast carcinoma in adult relatives.48,53 It is also established that these syndromes are largely associated with germline mutations of the p53 tumor suppressor gene. 54 There is also an association of the RMS with Beckwith-Wiedemann syndrome, which is associated with abnormalities on 11p15, where the insulin-like growth factor II gene is located.48,54 Associations with Costello’s syndrome are also described – an autosomal dominant inheritance syndrome, also associated with an increased risk of rhabdomyosarcoma. 55 Neurofibromatosis type 1 (NF1) and Noonan syndrome have also been reported in association with an increased risk, underscoring the partly genetic nature of the disease. 54
The processes of carcinogenesis remain unclear. 53 Three main types of RMS have been differentiated: (1) embryonal (ERMS): the most common, located mainly in the head and neck, as well as on the genitalia or urinary tract; (2) alveolar (ARMS): observed mainly in the arms or legs, chest, abdomen, or anogenital area; and (3) anaplastic (pleomorphic) (PRMS): an extremely rare variant characteristic of older age groups.54,55
RMS is localized mainly on the labia and rarely on the clitoris. 53 It clinically presents as a non-specific tumor mass, accompanied by vaginal bleeding or micturition disturbance in locally advanced cases.49,56 Usually adenocarcinomas of Bartholin’s gland and other cancers are considered in the differential diagnosis. 57 The histological findings are typical, including diffuse proliferation of rhabdoid cells that are round or polygonal in shape with eccentric nuclei, prominent nucleoli, and glassy eosinophilic cytoplasm containing hyaline-like inclusion bodies. 54
Embryonal rhabdomyosarcoma is the most common subtype, and presents mainly in early childhood. 53 There is an approximately equal frequency in both sexes, and their location in the genital area is second only to the head and neck.47,55
Alveolar RMS is very rarely localized on the vulva, with only single cases being reported. 58 The onset of ARMS is typically between 15 and 25 years, with predilection for the deep soft tissues of the extremities.58,59 Vulvar location accounts for less than 1% of all cases. 59
Aspiration biopsy of such tumors is characterized by the pattern of a small round cell tumor with predominantly single cells or stripped nuclei; cells are mainly uniform with rounded or irregular nuclei and scanty cytoplasm, and alveolar or rosette-like structures may also be present.59,60 Specific chromosomal aberrations are characteristic of ARMS, such as translocations of chromosome 2 and 13 [t (2; 13) (q35; q14)]. 61 According to recent studies, PAX3-FKHR fusion protein plays a major role in the pathogenesis of alveolar RMS. 61
Immunohistochemical expression of desmin confirms the diagnosis. 60 Small blue round cell tumors like Ewing’s sarcoma, peripheral primitive neuroectodermal tumor, poorly differentiated adenocarcinoma, lymphomas, and neuroblastoma must be considered in the differential diagnosis. 57
Botryoid rhabdomyosarcoma, also called sarcoma botryoides or botryoid sarcoma, is a variant of embryonal rhabdomyosarcoma, typical of ages below 8 years (average, 3 years), but there have been reported cases in older women as well.62,63
The name comes from the similarity of the clinical characteristics to ‘grape bunches’ – botryoid in Greek. 53 In cases of vulvar or vaginal localization, the objective symptom is a non-specific vaginal bleeding associated with the tumor mass. 61 The clinical picture can mimic a polyp or other neoplasm, and the most common localization is the labium majus. 63
The diagnosis of RMS is based on the histological features and additional diagnostic methods such as fine needle aspiration biopsy with subsequent cytological analysis and immunocytochemical staining for markers of myogenic differentiation.51,64 Expression of nuclear transcription factor myogenin (myf4) and MyoD1, myogenic transcriptional regulatory proteins that are highly specific markers for the identification of skeletal muscle differentiation, is characteristic of these tumors. 51 It should be borne in mind that, in rare cases, focal immunoreactivity to myogenin can be seen in fibroepithelial polyps, which can lead to diagnostic mistakes. 60
Carcinocythemia, representing the leukemic phase of a solid tumor, has been reported in isolated cases in association with alveolar rhabdomyosarcoma. 65 It is believed that the development of carcinocythemia is a bad prognostic sign, with a median survival after diagnosis of 8.5 weeks. 65
Local invasion, regional lymph node spread, and therapeutic response are important prognostic markers in RMS.51,61 Factors associated with a favorable prognosis are embryonal/botryoid histology, tumor size < or = 5 cm, with no metastases and age below 10 years at the time of diagnosis. 66 ). Besides locally infiltrative growth for RMS, lymphatic as well as hematogenous metastases to the bone marrow and lungs are characteristic. 52 Metastases have been rarely observed in the brain, liver, or spleen. 57 Recurrences are possible, and distant metastases are observed in approximately 30% of cases. 51
Multimodality therapy is the optimal therapeutic approach, but it is often associated with significant acute toxicities, especially during childhood. 67 The therapeutic aim is to achieve optimal treatment with minimal toxicity, preserving the quality of life in young patients.67,68 The more conservative approach – surgical excision of the primary tumor with efforts to minimize the resulting anatomic defect – may be preferable, but this is often difficult to achieve due to infiltrative tumor growth. 68 In cases where complete resection is not possible, radiotherapy is recommended. 68 Chemotherapy is mandatory in all cases of RMS. 69 Chemotherapeutic agents used with good effect are vincristine and dactinomycin with either cyclophosphamide (VAC) or ifosfamide (IVA).
Innovative therapeutic approaches include the use of topotecan or irinotecan, in combination with VAC.51,70 With their application, however, acute or chronic toxic effects can be observed, including myelosuppression, febrile neutropenia, hepatopathy, infertility, and second malignant neoplasms.71,72 Local therapy, consisting of surgery, brachytherapy (BT), and external-beam radiotherapy, may be administered in case of relapse. 73 In the case of non-metastatic tumors, conservative therapy with chemotherapy or radiotherapy after the initial excision has been reported with excellent results.73,74 Prognosis in non-metastatic RMS is very good, with an average 5-year survival of 85–90%.53,55,74
Pleomorphic rhabdomyosarcoma (PRMS) is a rare variant of the disease, thought to be extremely rare outside childhood. 75 It is usually localized in the lower extremities, abdomen/retroperitoneum, chest/abdominal wall, spermatic cord/testes, vagina, and vulva, and rarely in the upper extremities, mouth, and orbit. 75 Clinically, the tumor presents as a non-specific, asymptomatic mass, with sizes in the range of 1.5–15.0 cm (average, 7 cm). 76
The histological appearance consists of varying numbers of large, atypical, pleomorphic, polygonal rhabdomyoblasts, with abundant eosinophilic cytoplasm and a background of rounded or spindled rhabdomyoblasts.75,76 They differ from: (1) the classical form, by the predominance of atypical pleomorphic rhabdomyoblasts; (2) round cell PRMS, in which the characteristic finding is the presence of clusters of slightly atypical, medium-sized, round blue RMB; and (3) spindle cell PRMS, which is dominated by atypical spindled RMB arranged in a storiform growth pattern. 76
PMRS has an aggressive course, high metastatic potential, high mortality (>70%), and poor prognosis. 76
The spindle cell variant of rhabdomyosarcoma represents a rare variant of RMS, typical of childhood, with a more favorable course and prognosis. 77 Single cases in adults aged 18–79 years (average, 32 years) have been described with different clinicopathological characteristics from those typical of childhood. 78 Men are more commonly affected than women, and there is predilection for the head and neck. 77
Vulvar location is extremely rare, with only a single case reported. 79 Histologically, these tumors show long fascicles of spindled cells with elongated, vesicular nuclei and pale indistinct cytoplasm, as well as scattered spindled or polygonal rhabdomyoblasts with abundant brightly eosinophilic cytoplasm.79,80 Usually there is immunoreactivity to desmin, myf-4, fast myosin, myoglobin, HHF-35, and rarely to SMA.80,81 The literature has described isolated cases of spindle cell rhabdomyosarcoma of the vulva with myofibroblastic differentiation. 78
Conclusion
Sarcomas of the vulva are rare malignant neoplasms that often lead to misdiagnosis. They are characterized by non-specific clinical manifestations, aggressive behavior, and high metastatic potential and mortality. Prognosis is poor, and depends mainly on the size of the primary lesion, tumor invasion, and mitotic activity. Lesions greater than 5 cm in diameter, with infiltrating margins, extensive necrosis, and more than five mitotic figures per 10 high-power fields, are associated with an even worse prognosis, indicative for possible recurrence after surgical resection.
It is important to consider vulvar sarcomas in the clinical differential diagnosis of non-specific vulvar lesions, in order to establish an early accurate diagnosis and appropriate treatment.
Footnotes
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.