
Abstract
Background
Lysine methyltransferase 2 (KMT2) family proteins methylate lysine 4 on histone H3 (H3K4) to promote genome accessibility and transcription. Dysregulation or mutation of KMT2 family have been observed frequently in various types of human cancers. Colorectal cancer is the third most common cancer worldwide. However, few studies have evaluated the role of KMT2 family mutations in colorectal cancer. The present study aimed to explore the impact of KMT2 family mutations on clinicopathological, molecular characteristics and prognosis in colorectal cancer.
Methods
A total of 316 colorectal cancer patients were enrolled; tumor tissue and matched peripheral blood samples were collected and subjected to targeted sequencing with a panel of 1021 cancer-related genes. The association of clinical pathological features and molecular characteristics in patients were then analyzed. The cBioPortal dataset was used for investigating the KMT2 family mutations data and their correlation with clinical outcomes.
Results
The overall mutation frequencies of KMT2A-D were 9.5%, 0.5%, 13%, and 13%, respectively, which were more often present at right-sided primary and earlier stage tumors. KMT2A-D mutations are associated with enhanced genomic instability, including a higher level of microsatellite instability (MSI-H) and tumor mutational burden (TMB-H). In addition, our results highlight the co-occurring gene mutations within the Wnt signaling, ERBB2/4, TGF-β superfamily pathway, and PI-3-kinase pathway in KMT2-mutant colorectal cancer. KMT2 family mutations were predictive biomarker for better overall survival in metastatic colorectal cancer.
Conclusions
Collectively, we identified that KMT2 family mutations were correlated with higher-TMB and higher-MSI, thus resulting in a better outcome for colorectal cancer patients.
Introduction
The lysine methyltransferase 2 (KMT2, initially named as mixed lineage leukemia (MLL)) family members contained three subgroups, including KMT2A/KMT2B (sometimes called MLL4), KMT2C (also known as MLL3)/KMT2D (sometimes called MLL2), and KMT2F/KMT2G, encoding enzymes which transfer methyl groups on histone 3 lysine 4 (H3K4).1,2 Methylation of H3K4 is frequently associated with active transcription. 3 Thus, the KMT2 family plays critical roles in modulating genome accessibility and chromatin structures. A disrupted KMT2 family may lead to transcriptional dysregulation that subverts cellular identity through aberrantly linking proliferation and migration programs in cancers. 4 Over the past several years, next generation sequencing (NGS) has been widely applied to identify the alterations of KMT2 family members in blood cancers and solid cancers. 5 Four members of the KMT2 family, KMT2A, KMT2B, KMT2C, and KMT2D, are among the most frequently altered genes in different cancer types.2,6 In colorectal cancer (CRC), KMT2C/2D mutations were tightly associated with higher microsatellite instability (MSI-H) status and increased tumor mutation burden (TMB). 7 However, further understanding of KMT2 family mutations in CRC and their impact on pathological, molecular features and prognosis remains largely unknown.
The immunotherapy has revolutionized the landscape of treatment strategies in various cancer types, which previously limited by the shortage of effective treatment options, including a subset of CRC patients. 8 However, only a minority of CRC patients can achieve durable responses from these treatments. Further establishing highly reliable biomarkers for immune checkpoint inhibitor (ICI)-based immunotherapy will be of utmost importance to maximize therapeutic benefit. 9 In the clinic, high programmed death ligand 1 (PD-L1), MSI, and TMB are three conventional biomarkers when considering immune-based treatment strategies.10-12 Recently, Miao et al. 7 reported that KMT2C/2D mutations were tightly associated with MSI-H and higher TMB. In addition, a cohort of CRC patients receiving ICI treatment from Memorial Sloan Kettering Cancer Center (MSKCC) (Nat Genet 2019, dataset from cBioPortal) with KMT2C/2D mutations showed longer overall survival (OS). 7 A meta-analysis of 418 patients from independent ICI-treated datasets across multiple cancer types observed significant enrichment of KMT2 family mutations in responding tumors. 5 A pan-cancer analysis of the KMT2 family revealed that the expression of all KMT2 genes was associated with immune infiltrates and the tumor microenvironment. 13 However, the relationship between KMT2 mutations and molecular profiles in CRC is unclear. Further understanding of clinicopathological, genetic features and prognosis of patients with KMT2 alterations may be crucial in future personalized treatment for CRC.
In this context, we performed this study to explore the impact of KMT2 family mutations on clinical parameters, landscape of mutations and molecular pathways in CRC. We further analyzed the prognostic significance of KMT2 family mutations in CRC population regardless of treatment options with metastatic CRC patients from MSKCC (Cancer Cell 2018, dataset from cBioPortal).
Methods
Patients and samples
A total of 316 CRC patients receiving surgery at the Department of Colorectal Surgery, First Affiliated Hospital of Guangxi Medical University, were enrolled in this study. All patients provided written consent for genetic analysis, and the protocol was approved by the Ethics Committee of First Affiliated Hospital of Guangxi Medical University. Tumor tissues and matched peripheral blood samples were collected and sent to GenePlus (Beijing, China) for genetic testing. Right-sided colorectal cancer (RCC) consisted of tumors arising from the cecum to the hepatic flexure of the transverse colon, while left-sided colorectal cancer (LCC) consisted of tumors arising from the splenic flexure to the rectum.
Identification of genomic alterations
Genetic profiling was performed using targeted next-generation sequencing (NGS) as previously described.14,15 In brief, peripheral blood lymphocytes (PBL) was separated with centrifugation. Genomic DNA from PBLs and tumor tissues were separately obtained using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) and sheared. Sequencing libraries were generated with the KAPA DNA Library Preparation Kit (Kapa Biosystems, Wilmington, MA, USA). Barcoded genomic DNA libraries were hybridized to a custom-designed panel containing 1021 cancer-related genes. 16 DNA sequencing was performed on a HiSeq 3000 instrument according to the manufacturer's protocol (Illumina, Inc., San Diego, CA, USA).
Targeted capture sequencing required a minimal mean effective depth of 500 × in tissue DNA. Genomic alterations, including single nucleotide variants (SNV), small insertions and deletions (Indels), copy number alterations (CNA), and gene fusions/rearrangements, were detected with GATK, MuTect (version 1.1.4) and BreakDancer, respectively. Germline variants in PBL DNA samples were identified according to the Single Nucleotide Polymorphism database (dbSNP). 17 For quality control, the somatic mutations in tumor tissues were further filtered with the following inclusion criteria: (a) variants occur at <1% of the population frequency in the 1000 Genomes Project (https://www.internationalgenome.org/) and the Exome Aggregation Consortium; (b) absent in paired germline DNA from PBLs; and (c) present in greater than or equal to five high quality reads (Phred score ≥30, mapping quality ≥30), and without paired-end reads bias. MSI status was defined as MSI-H or microsatellite stable (MSS) using MSIsensor (v0.2). Tumors with an MSI score ≥ 10 were classified as MSI-H. Patients harboring ≥20 mutations/megabases (Mb) were classified as TMB-H, while those with <20 mutations/Mb were TMB-low (TMB-L).
Statistical analysis
Metastatic CRC (MSKCC, Cancer Cell 2018) dataset was downloaded from the cBioPortal website (http://www.cbioportal.org/, accessed on 9 March 2021). The chi square test or Fisher's exact test was used to tell frequencies of genetic alterations in different groups. A Mann–Whitney U test was performed to analyze the difference in TMB between KMT2 mutant and wild-type samples. The Kaplan–Meier method was used to analyze survival. Statistical processing was performed with SPSS version 23 (SPSS Inc., Chicago, IL, USA), and P < 0.05 (two-sided) was considered significant.
Results
Clinical characteristics
Samples from 316 patients with CRC were studied in our cohort. Patients’ clinical and molecular characteristics are shown in Table 1. The overall mutation rate of KMT2 was 21.8%. The positive rate of KMT2B mutations was 0.9% (3/316), while the other three were comparable, with KMT2A 9.5% (30/316), KMT2C 13.0% (41/316), and KMT2D 13.0% (41/316). Age at diagnosis ranged from 16 to 89 years, with a median of 55.9 years. KMT2 mutations were more frequently observed in tumors obtained from younger patients (54 vs. 59 years old, P = 0.001). No significant difference with gender was found between KMT2-mutant and wild-type CRC tumors (P = 0.18); 5.7% (18/316) patients were initially diagnosed with stage I, 22.8% (72/316) stage II, 28.5% (90/316) stage III, and 43.0% (136/316) stage IV. The presence of KMT2A, KMT2C, KMT2D, or KMT2 mutations were strongly associated with early stages, especially in patients with stage Ⅱ, all P < 0.05. The CRC samples included 218 (69.0%) colon cancer and 98 (31.0%) rectal cancer, with a total of 234 (74.1%) RCC and 82 (25.9%) LCC, respectively. There were strong associations of KMT2 mutant samples with right-sided tumors and colon cancers, P < 0.001 and P = 0.002, respectively.
Association of KMT2 family mutations and patients clinicopathological factors.
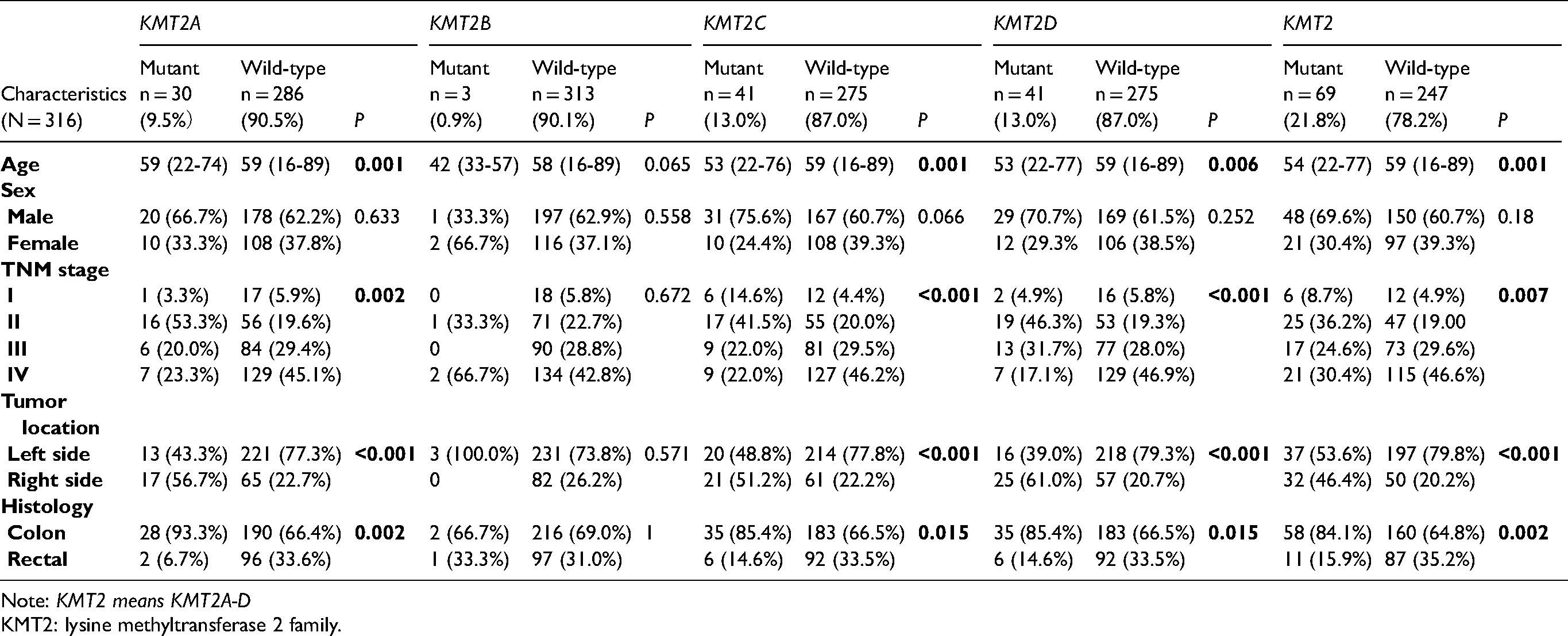
Note: KMT2 means KMT2A-D
KMT2: lysine methyltransferase 2 family.
Gene mutation profiles in the CRC cohort
With the application of targeted NGS, a total of 8093 non-silent somatic mutations (SNV and small Indels) in 1021 tumor-related genes were detected, with a median of 10 (from 0 to 409). The observed somatic mutations in CRC are shown in Figure 1. In our cohort, the most common mutations (≥10%) were found in TP53 (76.6%, 242/316), APC (75.3%, 238/316), KRAS (49.7%, 157/316), PIK3CA (20.9%, 66/316), LRP1B (20.3%, 64/316), TCF7L2 (19.9%, 63/316), FBXW7 (18.0%, 57/316), FAT2 (14.9%, 47/316), SMAD4 (14.9%, 47/316), ARID1A (14.2%, 45/316), KMT2C (13.0%, 41/316), KMT2D (13.0%, 41/316), FAM123B (12.7%, 40/316), BRCA2 (12.3%, 39/316), CTNNB1 (12.0%, 38/316), ATM (12.0%, 38/316), SOX9 (12.0%, 38/316), NOTCH3 (12.0%, 38/316), ERBB4 (10.8%, 34/316), and RNF43 (10.4%, 33/316).
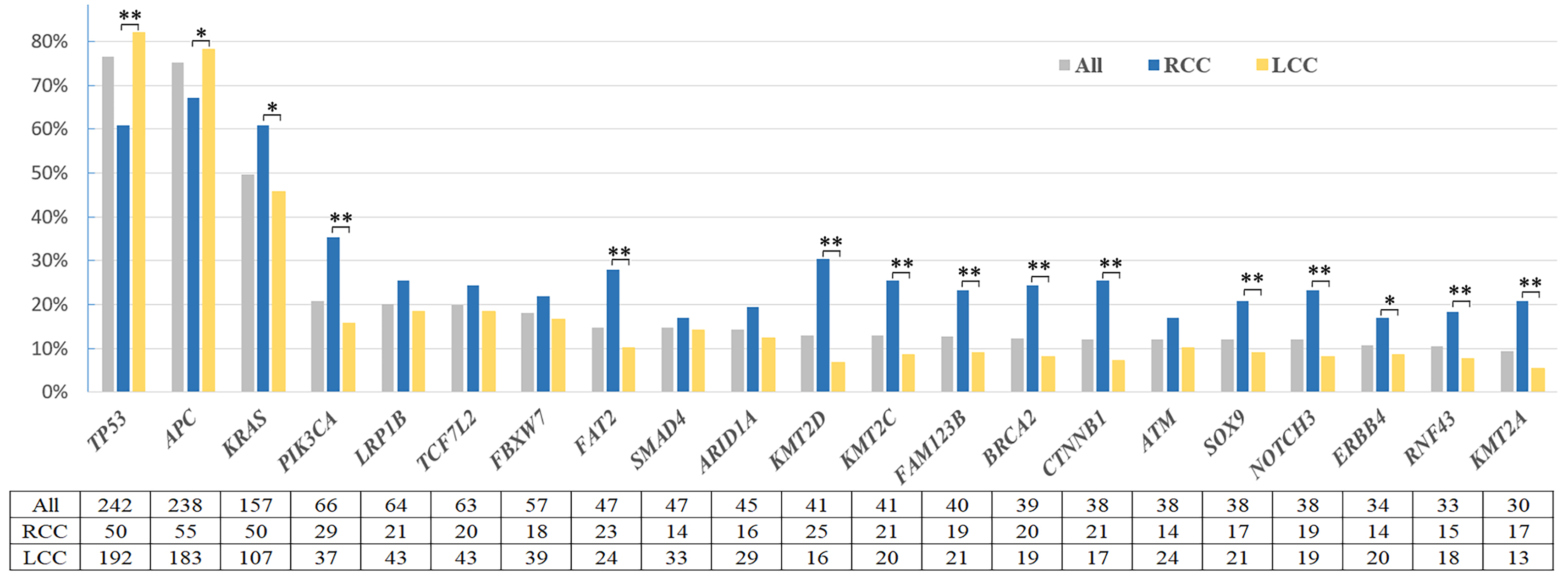
Frequency of common altered genes in right-sided and left-sided colorectal cancer.
The top two mutated genes with the highest mutation rates in CRC, TP53 and APC, presented with higher mutation rates in LCC when compared with RCC (both P < 0.05). In contrast, other most frequently mutated genes, KRAS, PIK3CA, FAT2, FAM123B, BRCA2, CTNNB1, NOTCH3, as well as KMT2C, KMT2D, and KMT2A, were significantly enriched in RCC patient samples (all P < 0.05).
Germline mutations were identified in 23 patients, mismatch repair (MMR) genes in 11 patients, and genes related to homologous recombination repair in 12 patients, including ATM, RECQL4, BRCA1, and others (Table S1).
Somatic mutation spectra of KMT2 family genes
Next, we focused on the mutational profile of four individual members in the KMT2 family (KMT2A, KMT2B, KMT2C, and KMT2D). As shown in Figure S1(a) to (d), the KMT2A-D proteins contain multiple domains, including Plant Homeodomain (PHD) finger, SET/Post-SET methyltransferase domain, FY-rich (FYR) regions, as well as domains that are specific for each member. 2 All KMT2 mutations detected are summarized in Table S2. Among those mutations, 19.0% (9/48) were truncating mutations and 81.3% (39/48) were missense mutations in KMT2A, while these were 24.4% (22/90) and 74.4% (67/90) in KMT2C, and 22.1% (19/86) and 76.7% (66/86) in KMT2D, respectively. No hotspot mutations were discovered in KMT2A/B/C/D. However, several common mutations of KMT2A/C/D were observed, for example, P773Rfs*8 (n = 2) in KMT2A, K2797Rfs*26 (n = 5) and F4496Lfs*21 (n = 5) in KMT2C and P2354Lfs*30 (n = 2) in KMT2D. Over 25% missense mutations were enriched around the N-terminus of KMT2C around the PHD region.
KMT2 family mutations with higher MSI and TMB status
We investigated the relationship of KMT2 mutations with TMB or MSI status. A total of 29 (9.2%) patients were MSI-H and 40 (12.7%) were TMB-H. Patients with either KMT2A/C/D mutation showed substantially higher MSI-H rate than wild-type patients, all P < 0.001 (Figure 2(a)). MSI-H and TMB-H rates were significantly higher in KMT2 mutant samples (39.1% and 55.1%, respectively) than in KMT2 wild-type samples (0.8% and 0.8%, respectively) (Figure 2(a) and (b)). Similarly, samples with either KMT2A/C/D mutation exhibited remarkably higher TMB levels compared with corresponding wild-type samples, all P < 0.001 (Figure 2(c)). Apart from MSI-H patients, 287 patients were found to be MSS. Higher TMB-H rates and TMB levels were also observed in MSS patients with either KMT2A/C/D mutation (all P < 0.001; Figure 2(d) and (e)).
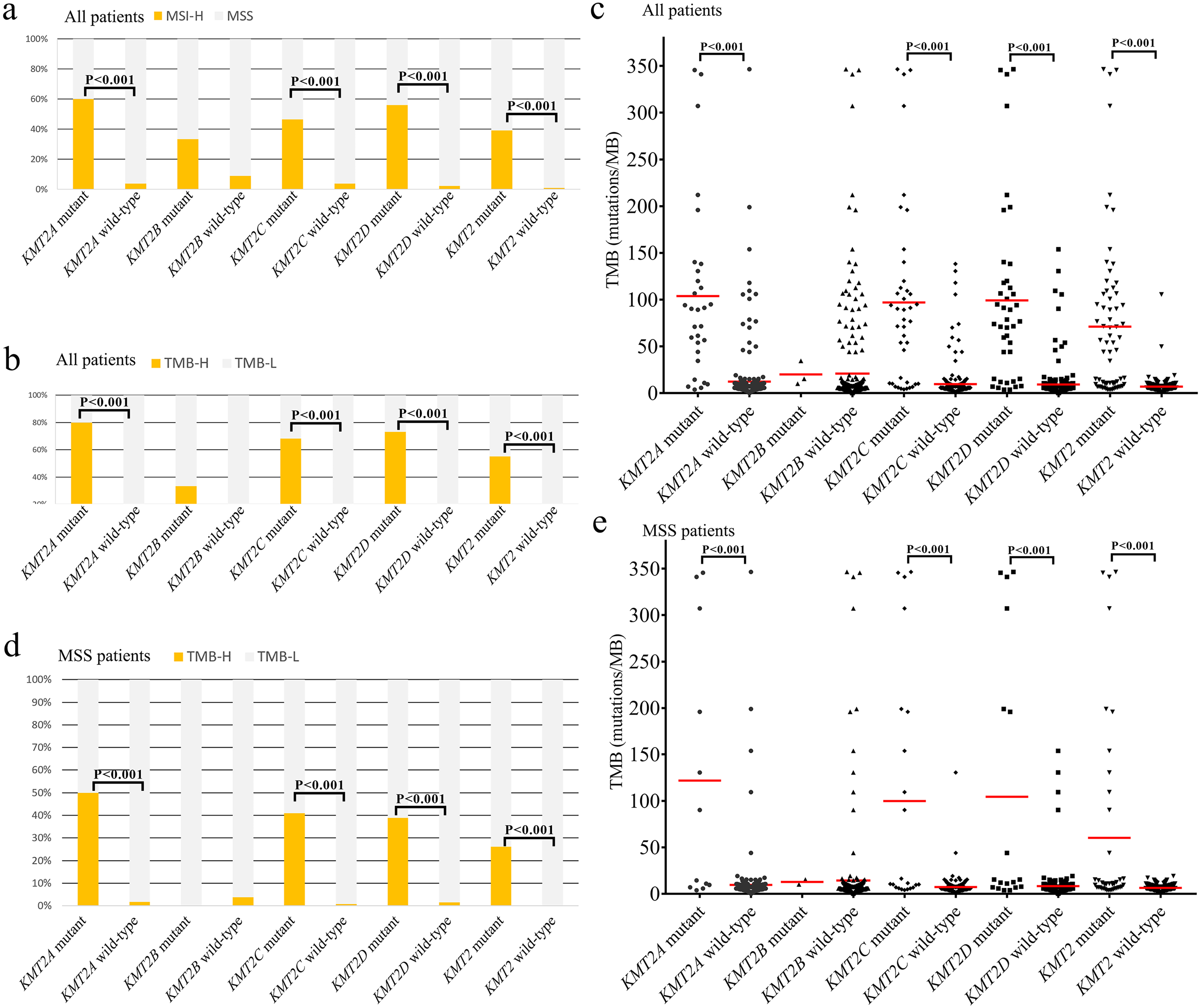
Association of KMT2 mutation and TMB and MSI status. (a) MSI status in all samples with KMT2A-D mutations. KMT2 mutant means at least one mutation of KMT2A-D was detected. KMT2 wild-type means no KMT2A-D mutation was detected. (b) TMB-H status in all samples with KMT2A-D mutations. (c) TMB in all samples with KMT2A-D mutations. (d) and (e) TMB-H and TMB in MSS samples with KMT2A-D mutations.
Co-occurring mutations, signaling pathway between KMT2 mutated and KMT2 wild-type tumors
Different KMT2 status showed different patterns of co-occurring mutations. We observed that co-occurring mutated cancer genes were specifically involved in five canonical signaling pathways: Wnt/β-catenin, RTK-RAS, P53, TGF-β, and PI-3-kinase pathway (Figure 3). Further, we found that the alteration frequencies of co-occurring genes in Wnt/β-catenin pathways (KMT2 wild-type vs. mutation) were FBXW7 (14.2% vs. 31.9%, P = 0.001), SOX9 (8.5% vs. 23.2%, P = 0.001), ARID1A (6.5% vs. 40.6%, P < 0.001), CTNNB1 (4.9% vs. 39.1%, P < 0.001), AXIN1 (3.3% vs. 13%, P = 0.004), and TCF7L2 (15.0% vs. 37.7%, P < 0.001). Alterations in the RTK-RAS pathway were ERBB4 (4.5% vs. 34.8%, P < 0.001), ERBB2 (2.4% vs. 26.1%, P < 0.001); SMAD2 (4.5% vs. 14.5%, P = 0.011), ACVR1B (1.6% vs. 15.9%, P < 0.001), TGFBR2 (2% vs. 15.9%, P < 0.001), TGFBR1 (0.8% vs. 10.1%, P < 0.001) were found in the TGF-β signaling pathway. Alterations in the PI-3-kinase pathway were PIK3CA (12.6% vs. 50.7%, P < 0.001), PIK3R1 (4.1% vs. 26.1%, P < 0.001), PTEN (3.7% vs. 21.7%, P < 0.001), and IGF1R (2.4% vs. 18.8%, P < 0.001). There were two alterations of p53 pathway members: ATM (5.3% vs. 34.8%, P < 0.001) were remarkably higher in the KMT2 mutated CRC analyzed, whereas TP53 (58.3% vs. 52.2%, P < 0.001) was enriched in KMT2 wild-type samples.
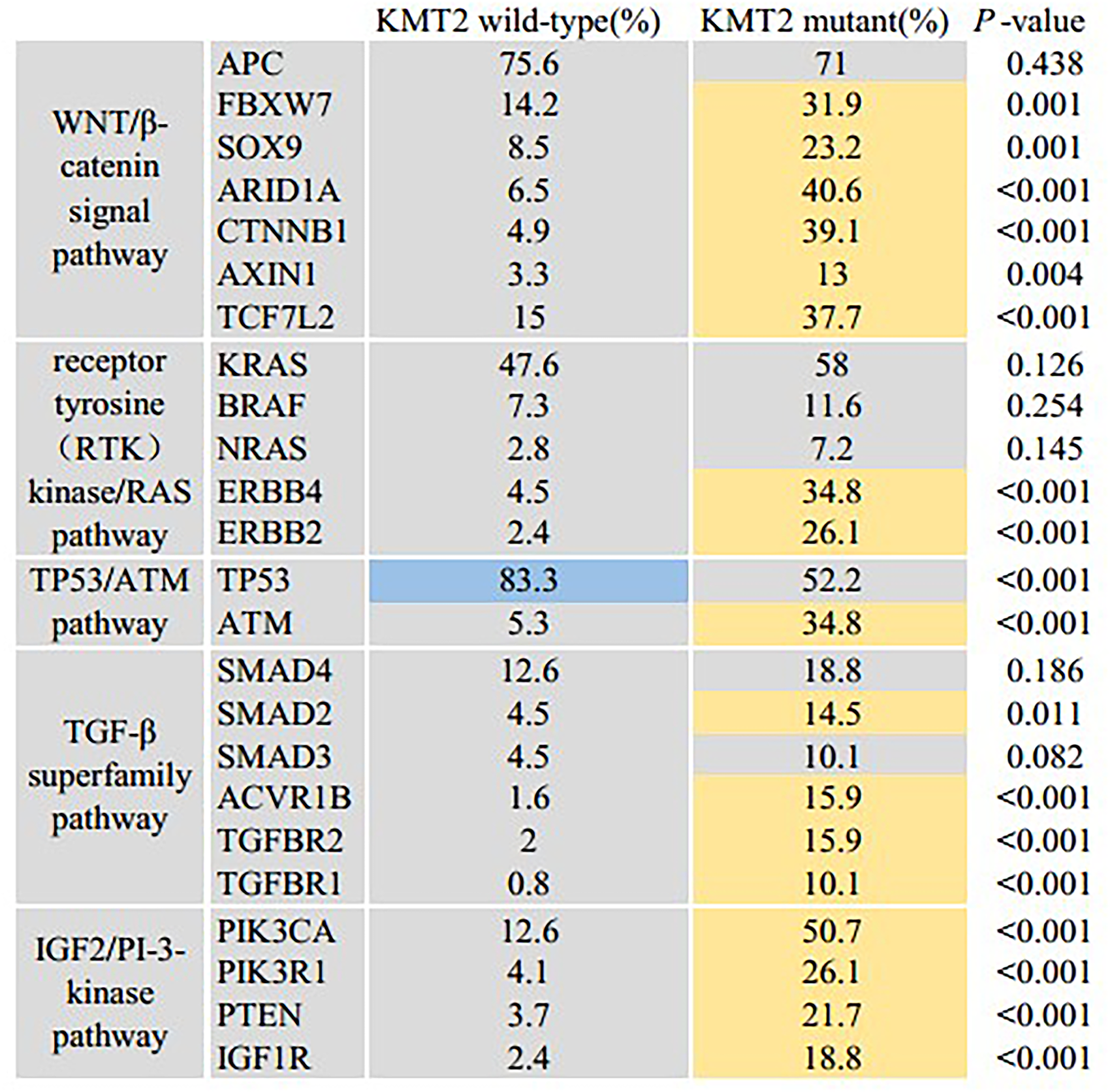
Significant differences of mutant genes between KMT2 wild-type and KMT2 mutant samples in five signaling pathways.
Clinical outcomes of KMT2 mutations in CRC
To better understand the outcomes of KMT2 mutated CRC cancers, we obtained the clinical information including KMT2 family mutations, MSI status, and survival in 1134 samples from 1099 metastatic CRC cases (MSKCC, Cancer Cell 2018), which was available at the cBioPortal website. This study included 597 male and 502 female patients. KMT2B was not involved in this cohort. The mutation frequencies of KMT2A, KMT2C, and KMT2D were 5.2%, 6.0%, and 9.4%, respectively. Similar to the results in our cohort, members of KMT2 mutations were significantly enriched in MSI-H samples (all P < 0.001; Figure 4(a)). OS analysis of KMT2 family mutations showed that wild-type patients presented moderately lower OS compared to KMT2-mutated patients (P = 0.056). The median OS (mOS) of KMT2 wild-type patients was 69.8 months, while the mOS in KMT2-mutated cases had not reached (Figure 4(b)). In 677 MSS patients, no significant difference between mOS between KMT2 mutant and wild-type patients was observed (72.7 months vs. 56.5 months; P = 0.390; Figure 4(c)).
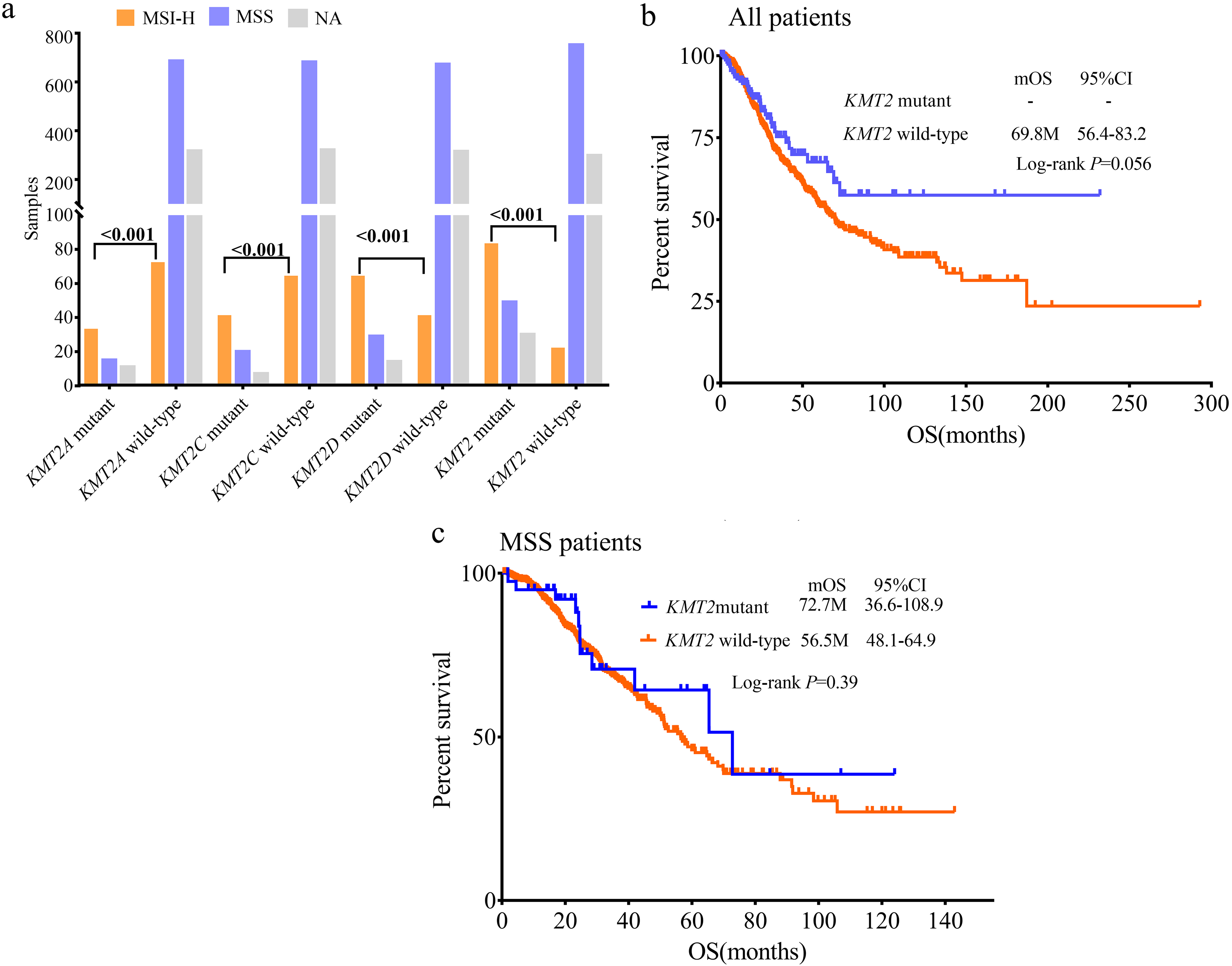
KMT2 family mutation feature and survival in MSKCC dataset. (a) Distribution of MSI status in KMT2A, KMT2C, KMT2D and KMT2 mutant samples. KMT2 mutant represents samples with at least one mutation in KMT2A, KMT2C or KMT2D. KMT2B was not involved in gene panels used in MSKCC research. (b) OS between patients with or without KMT2 mutation in all patients. (c) OS between patients with or without KMT2 mutation in MSS patients.
Discussion
In this study, we provide for the first time the impact of KMT2 mutations on molecular characteristics and survival in CRC. With 316 cases from our cohort and 1134 clinical samples from the MSKCC dataset, we found that KMT2 family mutations showed significant association with MSI-H and higher TMB levels, even in MSS samples. It was also highly associated with younger age, earlier tumor stage, and right-sided primary tumor location. We determined that KMT2 family mutant tumors mainly had co-occurring gene mutations related to Wnt signaling, TGF-β superfamily pathway, and PI-3-kinase pathway. In addition, mCRC with KMT2 family mutations had better OS compared with wild-type patients. Our findings suggested the possible role of KMT2 family mutations in the personalized treatment strategy in CRC patients.
Members of the KMT2 family play pivotal roles in methylating H3K4 to modulate genome accessibility and transcription. 2 In recent years, multitude exome-sequencing studies have revealed that KMT2 family genes are commonly mutated in a large variety of solid and blood malignancies, which were strongly linked to tumorigenesis, and immunogenicity. Accumulating evidence showed that KMT2D-mutant cells exhibited increased DNA damage, mutation burden, and interferon (IFN)-γ-stimulated antigen presentation. 18 Therefore, we highlighted the association between KMT2 family mutation and favorable prognostic biomarkers (MSI-H and TMB-H) for immune checkpoint inhibitors in CRC. Importantly, even KMT2 mutant samples with MSS status were associated with higher TMB. Recent data have showed that KMT2D-mutation sensitized tumor cells to immune checkpoint blockade (ICB) by promoting tumor immunogenicity in vitro. 18 A pooled study concerning KMT2 mutation on ICT clinical response, with 418 ICB-treated patients from all the publicly available studies, showed that clinical benefit of ICB was more prominent in the KMT2-mutant group compared with the KMT2 wild-type group. 5 A recently published large-scale analysis showed that gastric cancer patients with KMT2 mutations may also benefit from ICIs. 19 Further studies regarding the impact of KMT2 family mutant genes in CRC immune checkpoint therapy are needed.
Public disease-associated mutation databases (e.g., TCGA, COSMIC) have confirmed the KMT2 family as tumor suppressor genes in humans because cancer-associated truncating/inactivating mutations are widely dispersed in KMT2A-D genes. In our cohort, a total of 48 KMT2A mutations, 90 KMT2C mutations, and 86 KMT2D mutations in 316 samples were discovered—almost all these mutations were truncating/missense mutations. No hotspot mutations were discovered in the KMT2A/B/C/D genes. Over 25% of the mutations were enriched around the N-terminus of KMT2C. Whether the N-terminal region of KMT2C is hotspot region? Wang et al. 20 found that with the use of MutClustSW, missense mutation “hotspots” of KMT2C were strongly associated with the first PHD finger cluster and nearby region. In our cohort, over 25% KMT2C missense mutations were located around the first PHD finger cluster region. Learning more about the influence of these missense mutations on the biological function of the KMT2 family may help to uncover functionally important domains.
Many studies have shown the most recurrently altered genes between the left-sided and right-sided CRCs.21,22 In agreement with previous reports, KMT2A/C/D were enriched in the right-sided primary tumor location. 23 A study involving CRCs in adolescents and young adults showed that KMT2D significantly more commonly mutated in right-sided tumors. 24 Furthermore, we found that KMT2 mutations were more common in patients with earlier stages. Similarly, another gene, ARID1A, which responsible for chromatin remodeling, was also closely associated with right-sided primary and earlier stage tumors in CRC. 25 In addition, we compared the distinction of mutational characteristics of primary tumors in KMT2 mutant and KMT2 wild-type CRCs. We found that APC, KRAS, NRAS, and BRAF were commonly altered in CRCs regardless of KMT2 status, indicating their universally important role in CRC oncogenesis. TP53 mutations, which were reported to be moderately enriched in left-sided CRCs, were also significantly enriched in KMT2 wild-type patients. Co-occurring gene mutations related to Wnt signaling, ERBB2/4, the TGF-β superfamily pathway, and the PI-3-kinase pathway were significantly higher in KMT2 mutant samples. Consistent with our findings, a previous study showed that KMT2C and KMT2D mutations had significant co-occurrence with various driver mutations (e.g., PIK3CA, PTEN, and ARID1A) across multiple cancers, 6 suggesting that either KMT2C or KMT2D dysfunction individually might serve as a founder in the early stage of oncogenesis.
Several molecular features have been developed for prognostic stratification in CRC. The risk of recurrence was significantly lower for KRAS wild-type than KRAS mutant tumors in stage II colon cancer. 26 BRAF V600E mutations were closely related to inferior progression-free survival (PFS) and OS in MSS but not in MSI CRCs. 27 We found that the KMT2 family mutant mCRC exhibited a significantly shorter OS compared with KMT2 wild-type. While in MSS patients, KMT2 family mutations were related to moderate longer OS. Prolonged follow-up may help elucidate KMT2 family mutations in MSS and entire patients as the majority of patients in the dataset were alive. However, research in lung cancer demonstrated that the KMT2D mutation was significantly related to lower PFS and OS in non-small-cell lung cancer (NSCLC), and was not associated with survival in small-cell lung cancer (SCLC). 28 The opposite effects of the KMT2D mutation on survival between CRC and NSCLC is possibly due to the difference in tumor types.
Several limitations exist in this study: the retrospective nature of the analysis, the absence of treatment information and follow-ups in our group, and the lack of a DNA methylation evaluation. Future validation of current findings and use of multi-omics profiling of CRC in prospective and larger cohorts are needed.
Conclusions
In summary, we recruited 316 CRC patients and compared the clinical features and molecular characteristics between the KMT2 mutant group and the wild-type group in order to provide the preliminary insights of KMT2 mutations in CRCs. These findings reveal that the KMT2 family mutation may be a predictive biomarker for immune checkpoint inhibitors and prognosis.
Supplemental Material
sj-jpg-1-jbm-10.1177_03936155221095574 - Supplemental material for Correlation of KMT2 family mutations with molecular characteristics and prognosis in colorectal cancer
Supplemental material, sj-jpg-1-jbm-10.1177_03936155221095574 for Correlation of KMT2 family mutations with molecular characteristics and prognosis in colorectal cancer by Cun Liao, Wei Huang, Minglin Lin, Hui Li, Zihan Zhang, Xiaolong Zhang, Rongrong Chen, Mingfeng Huang, Pengli Yu and Sen Zhang in The International Journal of Biological Markers
Supplemental Material
sj-xlsx-2-jbm-10.1177_03936155221095574 - Supplemental material for Correlation of KMT2 family mutations with molecular characteristics and prognosis in colorectal cancer
Supplemental material, sj-xlsx-2-jbm-10.1177_03936155221095574 for Correlation of KMT2 family mutations with molecular characteristics and prognosis in colorectal cancer by Cun Liao, Wei Huang, Minglin Lin, Hui Li, Zihan Zhang, Xiaolong Zhang, Rongrong Chen, Mingfeng Huang, Pengli Yu and Sen Zhang in The International Journal of Biological Markers
Supplemental Material
sj-xlsx-3-jbm-10.1177_03936155221095574 - Supplemental material for Correlation of KMT2 family mutations with molecular characteristics and prognosis in colorectal cancer
Supplemental material, sj-xlsx-3-jbm-10.1177_03936155221095574 for Correlation of KMT2 family mutations with molecular characteristics and prognosis in colorectal cancer by Cun Liao, Wei Huang, Minglin Lin, Hui Li, Zihan Zhang, Xiaolong Zhang, Rongrong Chen, Mingfeng Huang, Pengli Yu and Sen Zhang in The International Journal of Biological Markers
Supplemental Material
sj-docx-4-jbm-10.1177_03936155221095574 - Supplemental material for Correlation of KMT2 family mutations with molecular characteristics and prognosis in colorectal cancer
Supplemental material, sj-docx-4-jbm-10.1177_03936155221095574 for Correlation of KMT2 family mutations with molecular characteristics and prognosis in colorectal cancer by Cun Liao, Wei Huang, Minglin Lin, Hui Li, Zihan Zhang, Xiaolong Zhang, Rongrong Chen, Mingfeng Huang, Pengli Yu and Sen Zhang in The International Journal of Biological Markers
Footnotes
Contributions
(a) Conception and design: C Liao, RR Chen, PL Yu; (b) Administrative support: C Liao, S Zhang; (c) Provision of study materials or patients: W Huang, ML Lin, H Li, ZH Zhang, XL Zhang; (d) Collection and assembly of data: C Liao, PL Yu; (e) Data analysis and interpretation: C Liao, S Zhang; (f) Manuscript writing: All authors; (g) Final approval of manuscript: All authors.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval
Analysis of the cases in this study were approved by the Ethics Committee of the First Affiliated Hospital of Guangxi Medical University.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.