
Abstract
Introduction
Recurrent Painful Ophthalmoplegic Neuropathy, previously known as Ophthalmoplegic Migraine, is a poorly characterized disorder mainly because there are few cases described. We report a new case of Recurrent Painful Ophthalmoplegic Neuropathy and a review of the literature to contribute to increasing the knowledge of the clinical features of this disorder.
Case report and review of literature
A 45-year-old woman presented with adult-onset recurrent attacks of abducens and oculomotor palsy associated with diplopia followed by headache. Most notably, pain always presented many days after oculomotor impairment, a feature never described in the literature. A diagnosis of possible Recurrent Painful Ophthalmoplegic Neuropathy was made after excluding other possible mimicking disorders. Symptoms usually resolved gradually with corticosteroid therapy, albeit without a clear-cut benefit.
Clinical data collected from 1989 to 2022 showed that adult onset in Recurrent Painful Ophthalmoplegic Neuropathy is not uncommon. While III cranial nerve palsy is typical, VI and IV nerve palsy have also been described.
Pathophysiology and diagnosis
Several hypotheses have been proposed, including nerve compression, ischemia or inflammation/demyelination, but none has been completely accepted.
Diagnosis remains of exclusion; magnetic resonance imaging and blood exams are key in differential diagnosis.
Conclusions
Our case gives us the possibility to expand the clinical features of Recurrent Painful Ophthalmoplegic Neuropathy, also contributing to updating the pathophysiological hypotheses.
Keywords
Introduction
Recurrent Painful Ophthalmoplegic Neuropathy (RPON), previously known as Ophthalmoplegic Migraine (OM), is a rare disease characterized by unilateral headache attacks associated with ocular cranial nerve palsy.
Since Charcot’s first formal description at the end of the 19th century, the advent of MRI provided significant insight into RPON; nonetheless, consensus concerning its pathophysiology, clinical characterization and optimal treatment strategies is still lacking, mainly because of the paucity of cases described.
The description of a new case gives us the opportunity to review the clinical features of RPON described in the scientific literature, which could be relevant to update the current pathophysiological hypotheses, also providing new insight into the appropriate treatment and prognosis of this rare and insufficiently known disease.
Epidemiology and clinical presentation: A review of cases
Our case
A graphic summary of this case is presented in Figure 1.

Time course of the symptoms complained of by our patient.
Our patient is a 45-year-old Italian woman who presented to our Clinic due to several episodes of diplopia associated with headache, nausea and vomiting.
Review of her medical history revealed gestational diabetes and breast cancer at 42 years of age, treated with quadrantectomy and subsequent radiotherapy; other than that, no chronic illness was found. The patient did not take nor was currently taking any medication chronically. Family history was negative for headache or neurological disorders in general.
She presented with a first episode of diplopia at 41 years of age, in the horizontal left and upward gaze. The patient was treated with prednisone 25 mg/day, starting seven days from the onset of diplopia, with reported resolution after several weeks.
Two years later, she presented with a new episode of diplopia with the same clinical characteristics, thus she immediately started treatment with prednisone 25 mg/daily, with benefit. However, 10 days later (upon tapering corticosteroid treatment), she presented with severe left retro-orbital pain, thus leading to corticosteroid reinstitution. The pain presented with a constant, stabbing quality and was associated with nausea and fatigue, partially responding to ibuprofen. No photo- or phonophobia were reported.
Angio-MRI and contrast enhancement brain MRI were performed, which were negative.
Pain improved but did not remit. Two months later, due to the reappearance of diplopia in the absence of corticosteroid therapy, the patient was admitted to an internal medicine ward, starting high-dose prednisone (80 mg iv) in the emergency room and 25 mg/day upon admission to the ward. During her stay, a rheumatological evaluation excluded giant cell arteritis, while a neurological one reported mild left abducens and trochlear nerve palsy. A lumbar puncture was then performed and cerebrospinal fluid (CSF) analysis, expanded to include oligoclonal bands, PCR for viruses, and cytological examination were negative. Autoimmune blood test screening showed positivity to ANAs (1:160 with nucleolar pattern). An ophthalmologic evaluation, comprising campimetry, was negative.
Corticosteroid therapy was subsequently tapered, with gradual resolution of both diplopia and headache in several weeks.
At 44 years of age, the patient presented with a new attack of diplopia, during which she came to the attention of our Clinic. Neurological examination confirmed diplopia in the horizontal left gaze. Physical examination was negative for other signs.
She was still treated with a corticosteroid cycle starting with prednisone 25 mg/day, with subsequent headache associated with nausea and fatigue during tapering. Similarly to the other episodes, diplopia reoccurred during tapering, prompting a new cycle with prednisone 25 mg/day which led to diplopia resolution but persistence of pain.
A brain MRI with focus on the cavernous sinus was repeated in order to exclude a diagnosis of Tolosa-Hunt syndrome, turning out negative, after which the patient was evaluated in our Clinic.
Blood exams, comprising full blood count, coagulation panels, glucose, sodium and potassium, liver and kidney function tests, total proteins, creatine phosphokinase (CPK), anti-ganglioside and onco-neural (anti-NMDAR, LGI1, CASPR2, DPPX, GABAB) antibodies only revealed a minimal positivity to anti-GD1b antibodies. CSF analysis, comprising the same exams as the former one, was repeated, still proving negative.
Pain partially responded to indomethacin; therapy with pregabalin (up to 150 mg/day) was proposed, but the patient refused because she had been free from attacks up to June 2022.
Described cases
Given its rarity, incidence of RPON is problematic to assess. In 1990, Hansen et al. (1) estimated it to be 0.7 cases per million inhabitants in Copenhagen County, but more recent studies to confirm this in other regions of the world have not been attempted.
Several works have tried to delineate a clinical picture of RPON: starting from the findings of two reviews from Gelfand et al. and Liu et al. (2,3), which stand out as the ones comprising the highest number of cases, we expanded the list by including cases described from 2020 to May 2022 (2–75): descriptive statistics and clinical features are provided in Table 1.
Clinical features of cases from references 2–72.
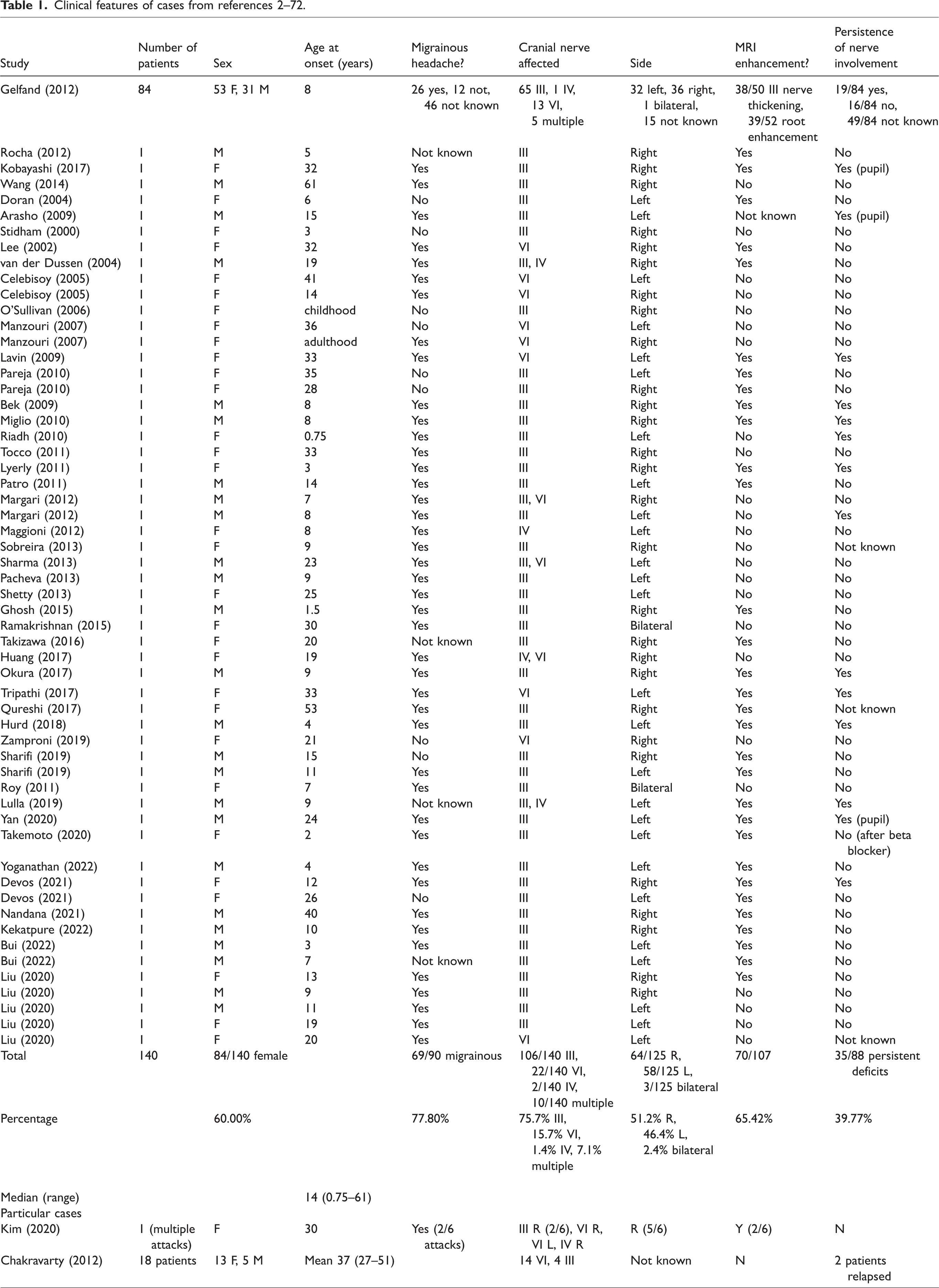
From current data, RPON appears to be a childhood or adolescence-onset entity, primarily affecting female patients. Headache shows migrainous qualities in the majority of cases; ophthalmoplegia strikingly affects the third cranial nerve, followed by the sixth; trochlear nerve involvement is seldom found in about half of cases associated with involvement of other nerves. More than half of cases show contrast enhancement consistent with the neuropathy. Corticosteroid therapy appears to be at least beneficial in most cases; persistence of cranial nerve involvement after the headache has subsided is not uncommon, sometimes only affecting pupil function.
In 2009, Lal et al. (76) published a case series of 62 patients, which showed significant differences in clinical presentation when compared to the majority of cases described by other authors. Most patients presented with adult onset and had abducens nerve involvement; most strikingly, neuroimaging was negative in all of them. Corticosteroid therapy was found to be beneficial.
These findings have led to propose that the entity described in this study is either a different disease or a subtype of RPON, as discussed for instance by Chakravarty et al. (58).
On a similar note, in order to explain the difference in contrast-enhancing and non-enhancing cases, Friedman (77) proposed dividing OM in primary or secondary entities according to contrast enhancement, on the hypothesis that absence of enhancement points to migraine while the contrary to neuropathy. This attempt has not been fully accepted as of now (58).
In their review, Liu et al. proposed incorporating contrast enhancement in MRI as a non-mandatory diagnostic criterion, as well as nerve distortion as observed in imaging (3).
Classification history and pathophysiology
During the 19th century, cases of recurrent oculomotor nerve palsy associated with headache were reported and attributed to various causes, such as nerve thickening due to neurosyphilis (2,78,79). A first formal definition was provided by Charcot in his 1890 study (80), referring to the entity as “migraine ophthalmoplegique”.
Since these preliminary descriptions, reports focused on providing a possible pathophysiological explanation to OM. While several hypotheses emerged, the common unifying mechanism was compression of the oculomotor nerves due to different mechanisms, including for instance aneurysmatic compression or pituitary edema (81–83).
In 1960, Walsh and O’Doherty postulated a compressive mechanism of disease in their case review, which employed arteriography (84): it was proposed that dilation of the internal carotid, basilar or posterior cerebral arteries compresses the oculomotor nerves during attacks. This hypothesis tried to link the migrainous aspect of OM to the ophthalmoplegic symptomatology. Also, this work attempted to distinguish primary cases from secondary forms due, for example, to lesions affecting cranial nerves, as reflected in the diagnostic criteria proposed by the authors, which would proceed to become the most accepted ones.
The compression hypothesis remained prevalent until 1980, when Vijayan reviewed the existing literature and provided evidence confronting it (85). First, arteriography was not positive in all cases; second, up to 67% of the reviewed patients did not present with significant pupillary abnormalities, which would be expected from extrinsic compression of the oculomotor nerve. Following instead the case of diabetic neuropathy, in which pupils are often spared due to the anatomical arrangement of parasympathetic fibers, the author then proposed an ischemic mechanism for OM, in which migraine-associated arterial swelling leads to delayed nerve ischemia and ophthalmoparesis.
The ischemic hypothesis was further corroborated by the finding of reversible thalamic ischemia ipsilateral to symptomatology in 2 OM patients by Shin et al. (6); nonetheless, it must be noted that the compression hypothesis has not been completely abandoned, as neurovascular conflict was detected in several, even recent, cases (85–86).
In 1988, the First International Classification of Headache Disorders (ICHD-1) was published, comprising OM as part of the migraine spectrum, even though the criteria did not specifically require headache with migrainous qualities (87).
A significant step forward was accomplished with the introduction of MRI, when Mark et al. first described reversible contrast enhancement and thickening of the affected nerves (88). This led to a third pathogenetic hypothesis, supported by the work of Lance and Zagami (15), according to which OM is caused by episodes of nerve demyelination due to different possible causes, such as inflammation or post-viral reaction. However, CSF in OM is typically unaltered and, except for an isolated case report associating cytomegalovirus immunoglobulin G (CMV IgG) to the disease (86), no particular associations with infections were ever demonstrated.
Nonetheless, a paradigm shift was occurring in which OM appeared as a neuropathy rather than a migraine: this was reflected in the second edition of ICHD, in which the disease, while retaining its original name, was moved to the cranial neuralgias and central causes of facial pain section (90). Curiously, criteria now required the headache to be migraine-like.
As already outlined in several reviews (2,89), the current debate focuses on whether RPON should retain an association with migraine or be definitely considered as a neuropathy. While current evidence points more to a neuropathic mechanism, a relationship to migraine cannot be completely excluded in several patients (89), as will be shown below.
In 2018, the third edition of ICHD changed the name of OM to RPON in order to reflect this view (91). Our case is significant as diplopia always preceded headache by a significant number of days, an instance which has never been described in the literature. Nonetheless, formal criteria for the diagnosis of RPON are respected; extensive testing, in particular repeated magnetic resonance imaging, has excluded other mimicking entities such as Tolosa-Hunt Syndrome, which, differently from RPON, strictly requires headache to precede oculomotor paresis in its diagnostic criteria as defined by the ICHD-3 (91).
Furthermore, we believe that the absence of any inflammatory or immune alteration in our case, save for the slight positivity to anti-GD1b antibodies (not deemed to be significant due to scarce specificity), is an element speaking against the inflammatory/demyelinating hypothesis of RPON.
Differential diagnosis
In suspected RPON, the first diagnostic question to address, especially in adult-onset cases, is ruling out a mimicking lesion. From a review of the literature it emerges that schwannomas are the most commonly implicated masses (92–96), followed by venous angiomas and hamartomas. Concerning the latter, Akimoto et al. proposed in their work that hamartomas may cause RPON through trigeminovascular-mediated vasodilation and subsequent nerve strangulation (97).
Being a rare entity, RPON can be easily mistaken for other, more common causes of painful ophthalmoplegia.
Moving from neoplastic etiologies, disorders causing painful ophthalmoplegia constitute a vast group with different causes, comprising inflammatory, vascular, infectious, and traumatic ones, as highlighted in Gladstone’s review (98). Among the various disorders, Tolosa-Hunt syndrome (THS), in which granulomatous inflammation of the cavernous sinus leads to corticosteroid-responsive painful ophthalmoplegia, stands out as a mimicker of RPON (99,100), especially in the rarest cases where MRI does not show typical features of THS.
In summary, diagnosis of RPON requires exclusion of other more common disorders: this can be achieved by laboratory testing on blood and CSF, ruling out inflammatory and infectious diseases, and neuroimaging with MRI excluding lesional, vascular or traumatic causes.
Interestingly, a work by Maggioni et al. suggested that electroencephalographic abnormalities are present during RPON attacks, even resembling patterns compatible with migraine with aura or complicated migraine attacks (44): this would not only be useful for diagnostic purposes but would also constitute evidence in favor of a migrainous nature of the disease.
Treatment and prognosis
Optimal treatment of RPON has not been elucidated: given its possible inflammatory etiopathogenesis, corticosteroids have been the most employed strategy, as outlined in literature reviews (2,3). Such treatment appears to benefit a consistent proportion of patients: however, given the scarcity of cases, blinded trials have not been devised.
Several other lines of treatment have been tried, including indomethacin in two patients (40), pregabalin after corticosteroid failure in one patient (57) and beta-blockers (67).
Concerning prognosis, ophthalmoplegia is typically reversible; however, persistent neurological signs were found in a small but significant proportion of patients (19,22). Treatment with botulinum toxin has been proposed to correct eye misalignment in these cases (101).
Conclusions
RPON is a disorder whose characterization is far from exhaustive. This is due to the rarity and likely the difficulty of recognizing and reporting cases. Even existing case reviews may sometimes lack details which would help better characterize this entity.
A main point of discussion is whether migraine should be associated with RPON: while accumulating evidence leans toward a neuropathic disorder, the significant percentage of patients presenting with migrainous headache cannot be overlooked. While MRI often shows alterations, the fact that not all patients present with contrast enhancement is another perplexing piece of the puzzle.
It might be possible that RPON might actually be OM, as first thought; still, it cannot be excluded that several different entities with different pathophysiology are mistaken as a single, unified disorder. Clinical characteristics of our case may contribute to expanding the clinical phenotype of this rare disorder suggesting a neuropathic etiopathogenesis.
Clinical implications
Recurrent Painful Ophthalmoplegic Neuropathy is a poorly understood disorder, whose pathophysiology still remains elusive We provide a case report with atypical features (adult onset, diplopia preceding headache) in which diagnosis has been made by exclusion More “atypical” cases might have been missed in the past While case reports are helpful in rare disorders such as RPON, they are probably not ideal in defining their characteristics.
Footnotes
Acknowledgements
The Authors wish to thank Cecilia Baroncini for her help in revising and providing corrections to the manuscript.
Ethical approval and patient consent
The patient provided written consent for publication of her case report.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.