
Abstract
Objective
To investigate the role of calcitonin gene-related peptide, pituitary adenylate cyclase-activating polypeptide-38 (PACAP38) and vasoactive intestinal polypeptide in cluster headache, we measured these vasoactive peptides interictally and during experimentally induced cluster headache attacks.
Methods
We included patients with episodic cluster headache in an active phase (n = 9), episodic cluster headache patients in remission (n = 9) and patients with chronic cluster headache (n = 13). Cluster headache attacks were induced by infusion of calcitonin gene-related peptide (1.5 µg/min) in a randomized, double-blind, placebo controlled, two-way cross-over study. At baseline, we collected interictal blood samples from all patients and during 11 calcitonin gene-related peptide-induced cluster headache attacks.
Results
At baseline, episodic cluster headache patients in remission had higher plasma levels of calcitonin gene-related peptide, 100.6 ± 36.3 pmol/l, compared to chronic cluster headache patients, 65.9 ± 30.5 pmol/l, (p = 0.011). Episodic cluster headache patients in active phase had higher PACAP38 levels, 4.0 ± 0.8 pmol/l, compared to chronic cluster headache patients, 3.3 ± 0.7 pmol/l, (p = 0.033). Baseline levels of vasoactive intestinal polypeptide did not differ between cluster headache groups. We found no attack-related increase in calcitonin gene-related peptide, PACAP38 or vasoactive intestinal polypeptide levels during calcitonin gene-related peptide-induced cluster headache attacks.
Conclusions
This study suggests that cluster headache disease activity is associated with alterations of calcitonin gene-related peptide expression. Future studies should investigate the potential of using calcitonin gene-related peptide measurements in monitoring of disease state and predicting response to preventive treatments, including response to anti-calcitonin gene-related peptide monoclonal antibodies.
Introduction
The hallmark of cluster headache (CH) is periodicity and prominent cranial autonomic symptoms (CAS) (1). Most patients report periodicity by experiencing episodic CH with month-long attack periods separated by remission periods (2). To what extent mechanisms underlying CAS contribute to initiation of CH attacks is still not fully elucidated. The trigemino-autonomic reflex activation is associated with release of sensory and parasympathetic neuropeptides (3) such as calcitonin gene-related peptide (CGRP), pituitary adenylate cyclase-activating polypeptide-38 (PACAP-38) and vasoactive intestinal polypeptide (VIP). Studies investigating plasma levels of these neuropeptides in CH remain, however, scarce and conflicting (4–8). Elevated plasma CGRP, VIP and PACAP38 have been reported during spontaneous and glyceryl trinitrate (GTN) provoked CH attacks (5–7,9). The diverging methodologies of these studies, however, make it difficult to compare findings across studies. To date, no studies have investigated the role of these vasoactive peptides in chronic CH patients. In addition, it is unknown whether plasma levels correlate to disease periodicity. As CH is unpredictable and short lasting, investigation of patients during spontaneous attacks can be immensely difficult. This challenge can be overcome by studying provoked CH attacks in a controlled setting (5). Recently, we demonstrated that CGRP infusion could provoke CH attacks in patients in an active disease state (episodic CH in active phase and chronic CH patients), but not in patients during remission (10).
We hypothesized that baseline levels of CGRP, VIP and PACAP38 would be elevated in episodic and chronic CH patients in an active disease phase compared to patients in remission. Furthermore, we hypothesized that CGRP-induced CH attacks would cause a further increase in plasma levels of neuropeptides. To test these hypotheses, we investigated plasma CGRP, VIP and PACAP38 at baseline and during CGRP-induced CH attacks. In addition, we compared the baseline concentration of neuropeptides in CH patients with historical data on migraine patients and healthy controls.
Materials and methods
Patients were eligible for inclusion if they were aged 18–65 years and had a verified diagnosis of episodic or chronic CH as defined by the International Headache Society classification (Headache Classification Committee of the International Headache Society (ICHD-3 beta), 2013) (11). We recruited participants from the outpatient clinic at the Danish Headache Center (Rigshospitalet-Glostrup) in the period from December 2015 to April 2017. The present study is a predefined part of a larger parent protocol (protocol H-15006836, clinicaltrials.gov identifier NCT02466334). The first part of the study investigated the ability of CGRP to induce cluster headache-like attacks and has previously been described in detail (10). Patients were eligible for inclusion in the study if they were in active disease phase, defined as occurrence of typical CH attacks within the last 30 days; or in remission, defined as attack-free for at least 30 days. Episodic patients could participate in remission and in active disease phase. According to the ICHD-3 beta criteria, chronic patients did not have attack-free periods exceeding 30 days in the last 12 months. Exclusion criteria included any other type of headache (apart from episodic tension-type headache ≤ 5 days per month), any previous serious somatic or psychiatric condition, pregnant or nursing women, drug misuse or daily intake of medication other than preventive treatment for CH. All patients underwent a full medical examination and, in women of childbearing age, pregnancy testing was conducted prior to participation.
The study was approved by the Regional Committee on Health Research Ethics of the Capital Region (H-15006836) and was conducted in accordance with Helsinki II Declaration of 1964, with later revisions. The study was registered at clinicaltrials.gov (identifier NCT02466334) and approved by the Danish Data Protection Agency. All patients received oral and written information about the study and were given time for consideration before giving their written consent to participate.
In the present study, we conducted post hoc analyses that included previously published data on migraine patients and healthy volunteers (12) (ClinicalTrials.gov identifier NCT01841827). All samples from previous data were collected in an interictal state, defined as the participant being completely headache and analgesic free for a minimum of 48 hours prior to sampling. Samples were analyzed by the same assays in the same laboratory as the current study (12).
Design and experimental protocol
The study was conducted as a randomized, double-blind, placebo controlled, two-way cross-over study. All patients were randomly allocated to receive a continuous infusion (Braun Perfusor, Melsungen, Germany) with either 30 µg CGRP (1.5 µg/min) (Calbiochem® and PolyPeptide group) or placebo (saline) over 20 min on two separate days. CGRP and placebo were prepared in identical vials and randomized by the regional central pharmacy. Allocation was balanced to ensure approximately even numbers of participants receiving CGRP first and placebo last, or vice versa. The randomization code remained in the hospital during the study and was unavailable to investigators until study completion.
On both experimental days, patients with episodic (active phase) and chronic CH reported themselves to the clinic when they were headache/attack free for at least 3 and 8 hours, respectively. An 8-hour headache-free-interval prior to provocation was initially set for both episodic patients in cluster and chronic patients, but due to feasibility concerns a revised 3-hour headache-free-interval was set in order to include episodic patients with a high mid-cluster attack burden.
All participants were asked to retrospectively estimate their attack frequency in the preceding 30 days. Patients were placed in a supine position and a venous catheter (Venflon®) was inserted in the cubital vein on the right or left arm for CGRP infusion and drawing of blood samples. Patients were at rest for 15 min before obtaining baseline status. Blood for analysis of CGRP, PACAP38 and VIP was drawn at fixed time points: At baseline (T0), post infusion (T20), 10 min (T30) and 70 min post infusion (T90). If the patient developed a CH-like attack during the observations period, blood was drawn at the onset phase of attack (Ta0), after 15 min (Ta15) and at 30 min after the start of the attack (Ta30).
Blood collection and processing
Blood was drawn through the venous catheter and connector using two 20 ml syringes. For blood sampling, the first 5 ml were discarded and after the procedure the catheter was flushed with saline. The blood was thereafter transferred into different tubes: Precooled lithium heparin tubes containing aprotinin (Trasylol®) for VIP; precooled EDTA tubes with aprotinin for PACAP38, and standard EDTA tubes for CGRP. All tubes were inverted several times. The precooled tubes were stored in a cooling box (5℃) and the rest stored at room temperature for 20 min until centrifugation. The tubes were centrifuged together at 4℃ at 1851 g for 10 min. Plasma was thereafter transferred to polypropylene tubes (Greiner Cryo.s™) and stored at −25℃ until analysis.
Radioimmunoassay
Plasma CGRP concentrations were measured with a fully evaluated radioimmunoassay for human CGRP, as described previously (Schifter, 1991) (12,13). The tracer was prepared by the method of Iodogeneral (Pierce, Rockford, IL, USA) (14) by iodination of [Tyr0] α-CGRP (25–37) amide and purification by high liquid chromatography (HPLC). Samples, antibody and calibrators were incubated at 4 ℃ for about 90 hours before addition of tracer and subsequent incubation for 48 hours. Free and antibody-bound tracer were separated by Sac-Cel separation.
Plasma concentration of PACAP38
The concentration of PACAP38 in plasma was measured radioimmunochemically using antiserum 733C-5 directed against the sequence PACAP28–38 (15). The antiserum that was used at a final titer of 1.2 × 105 in a total volume of 0.8 ml/tube does not cross-react with PACAP27, VIP, or other structurally related peptides. Synthetic PACAP28–38 labeled to a specific radioactivity of 30 Bq/mol with 125I by the iodogen method was used as tracer and synthetic human PACAP38 was used as standard. The IC50 value (the concentration of PACAP38 giving 50% displacement of the tracer) was 17 pmol/l, and the intra-assay and inter-assay coefficient of variation values were 3.1 and 10.1%, respectively.
Since PACAP38 in human plasma is bound to the protein ceruloplasmin (16), the peptide was freed from ceruloplasmin before measurement by the following procedure: 1.2 ml of 1% trifluoroacetic acid was added to an equal volume of plasma from each subject and mixed thoroughly for 60 s. After incubation for 10 min in an ice bath, the mixture was neutralized by addition of 15 μl of 5 M NaOH. Subsequently, 2.5 ml of absolute ethanol was added. After thorough mixing, followed by centrifugation at 1500 g for 20 min at 4℃, the supernatant was decanted and dried under vacuum. The dried product was reconstituted to its original volume with assay buffer for assay.
Plasma concentration of VIP
The concentration of VIP in plasma, after extraction with absolute ethanol, was measured by the VIP radioimmunoassay using antiserum 5603–6 at a final titer of 1.2 × 106 in a total volume of 0.8 ml/tube (17,18). This antiserum recognizes the mid- and C-terminal regions of the VIP molecule (sequence 11–24) and displays no cross-reactivity with other known gastrointestinal peptides or neuropeptides. The label has a specific radioactivity of 0.92 nCi/fmol (∼34 Bq/fmol). The IC50 value (the concentration of VIP giving 50% displacement of label) was 24 pmol/l and the intra-assay and the inter-assay coefficient of variation values were 8.7 and 12.6%, respectively.
Headache characteristics and vital signs
From baseline (T-10 and T0) and throughout the entire experiment the following variables were recorded every 10 min: Headache intensity on a verbal rating scale (VRS) from 0 to 10 (0; no headache, 1; very mild headache, 10; worst imaginable headache); quality of pain (stabbing, throbbing, pulsating or resembling usual CH attack); headache localization and accompanying symptoms, these including CAS. In addition, we recorded blood pressure and heart rate. Symptoms experienced outside recording intervals were documented separately.
Statistical analysis
All absolute values are presented as mean ± standard deviation. The primary endpoints were: a) Differences of biochemical variables (CGRP, PACAP38 and VIP) in between groups (episodic CH patients in active phase, episodic CH patients in remission and chronic CH patients) at baseline; b) differences over time in plasma concentrations of biochemical variables between patients developing an attack and those who did not; c) differences in plasma concentrations over time of biochemical variables between active and placebo days.
Distribution of demographical data was tested using the D'Agostino and Pearson normality test and group comparisons of demographical data were subsequently analyzed using parametric statistics. Evaluation of baseline variables was done using a generalized linear model with repeated measurements.
To analyze for an effect of CGRP infusion on biochemical variables, we used repeated measurements analysis with random effect of subjects, attacks and further of subjects times day. In this way, we allow for correlation between measurements on the same individual, and additional correlation between measurements in the same individual on the same day. The measurement taken at time zero was used as baseline variable in the repeated measurements model. For each of the responses, VIP, PACAP and CGRP, we checked model assumptions and transformed the response variables as appropriate to meet model requirements. Correlations between baseline levels and time since last attack were calculated using the Pearson correlation test.
We used GraphPad Prism 7.02, SAS Enterprise and R 3.4.3 for statistical analyses. All p-values were two-sided and considered significant if < 0.05.
Data availability
The data supporting the findings of this study are not publicly available, but will be shared, in an anonymized form, by request from any qualified investigator.
Results
In total, 31 patients (26 men and five women) completed the study (Figure 1). The mean age was 37 years, (range 19–59). Nine patients reported episodic CH in active phase (six men, three women; mean age 32, range 19–56 years), nine episodic in remission (all men; mean age 32, range 22–43 years), and 13 chronic CH (10 men, three women; mean age 42, range 26–59 years). Clinical data on patients are shown in Table 1. Episodic patients in remission reported remission on average for 6.6 (range 1.3–18.0) months prior to participation in the study. At baseline, blood samples were collected in all 31 patients for VIP and CGRP, but samples from one patient were lost for PACAP38. CGRP infusion induced a CH-like attack in 16 out of 31 patients, of these none were episodic patients in remission. We collected blood samples during 11 out of 16 CH attacks, and all attack samples were collected prior to abortive treatment. In the remaining five attacks, symptoms subsided before we had the chance to engage the attack sampling protocol. All provoked attacks were unilateral, located in the periorbital region, and were accompanied by CAS and/or restlessness. The median severity of provoked attacks was 10 (IQR 4–10, range 1–10) and median number of accompanying symptoms was four (IQR 1.5–5, range 1–8). Characteristics of 11 provoked attacks are listed in Table 2. The concentration of CGRP, PACAP38 and VIP were above the detection limit in all blood samples.
Flow chart of recruitment and inclusion of patients. Clinical data on patients with episodic cluster headache in active phase (eCHa), remission (eCHr) and chronic cluster headache (cCH). eCHa: episodic cluster headache patient in active phase; eCHr: episodic cluster headache patients in remission; cCH: chronic cluster headache patients; SD: standard deviation, W/M: Women/Men. Treatments: Verapamil 400 mg; verapamil 560 mg; verapamil 400 mg; recent blockade of greater occipital nerve. Treatments: Verapamil 400 mg; verapamil 800 mg. Treatments: Verapamil 440 mg; verapamil 480 mg; verapamil 100 mg; verapamil 400 mg and melatonin 8 mg, verapamil 240 mg, verapamil 600 mg; verapamil 240 mg and lithium 200mg; pulsatane SPG microstimulator system and melatonin 4 mg. Clinical characteristics of provoked attacks in 11 patients. Headache intensity: 0–10 verbal response scale. Ta0: Attack onset, prior to acute therapy. Ta15 and Ta30: 15 and 30 min after attack onset respectively. Acute therapy: Suma: sumatriptan 6 mg sc; Oxy: oxygen 15 L/min Optimask; SPG: the Pulsante SPG Microstimulator system; Dic: Diclofenac 25 mg sc. Accompanying symptoms: Lac: lacrimation; pto: ptosis; mio: miosis; con: nasal congestion; inj: conjunctival injection; swe: forehead and facial sweating; res: restlessness; rhi: rhinorrhea; ede: eyelid edema.


CGRP
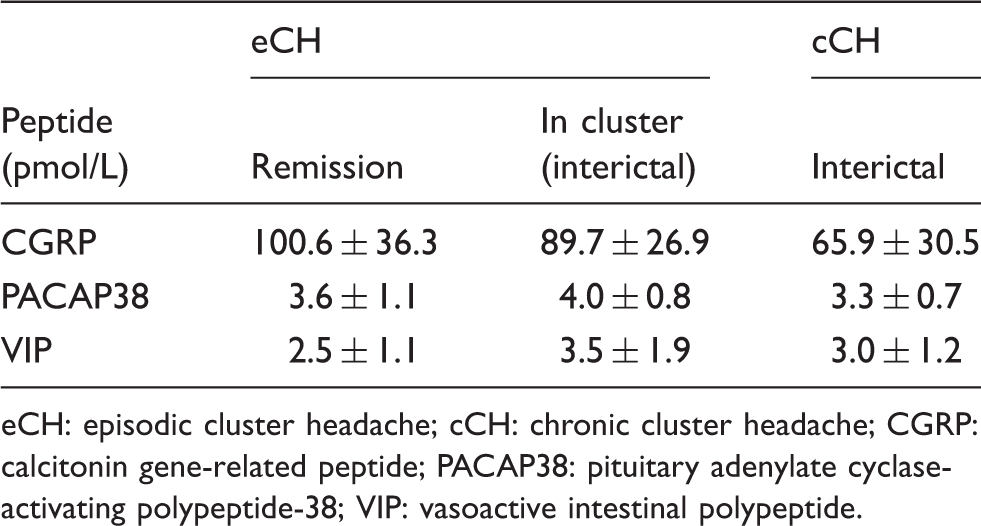
We found significantly higher baseline plasma CGRP in episodic patients in remission, 100.6 ± 36.3 pmol/l, compared to chronic patients, 65.9 ± 30.5 pmol/l, (p = 0.011) (Figure 2, Table 3). The repeated measurements analysis showed no independent increase of plasma CGRP in patients who reported a CH-like attack after provocation with CGRP (p = 0.36). After CGRP infusion, plasma levels of CGRP increased significantly, 574.3 ± 296.4 pmol/l, compared to baseline, 81.7 ± 33.4 pmol/l, (p < 0.0001). We found no changes in CGRP levels after placebo infusion compared to baseline (p = 0.43).
Baseline levels of CGRP, VIP and PACAP38 presented as means and SD. Levels of CGRP, PACAP38 and VIP at baseline, presented in means ± SD. eCH: episodic cluster headache; cCH: chronic cluster headache; CGRP: calcitonin gene-related peptide; PACAP38: pituitary adenylate cyclase-activating polypeptide-38; VIP: vasoactive intestinal polypeptide.
PACAP38
We found significantly higher baseline PACAP38 levels in episodic patients in active phase, 4.0 ± 0.8 pmol/l, compared to chronic patients, 3.3 ± 0.7 pmol/l, (p = 0.033) (Table 3). CGRP-induced CH attacks were not associated with changes in plasma PACAP38 (p = 0.29). Compared to baseline, plasma levels of PACAP38 remained unchanged after CGRP (p = 0.66) and placebo (p = 0.57) infusion.
VIP
We found no differences in baseline plasma levels of VIP between CH groups (p > 0.05) (Table 3). The repeated measurements analyses revealed a significant decrease in patients who reported a provoked attack (p = 0.013). Infusion of CGRP caused a significant increase in plasma VIP compared to baseline (p < 0.001), but not after placebo (p = 0.53).
Post hoc analysis
We compared five groups: Episodic patients in active phase; episodic patients in remission; chronic patients; migraine without aura patients and healthy controls (Figure 2). The analysis revealed that episodic patients in bout (p < 0.001), patients in remission (p < 0.001) and chronic patients (p = 0.020) had higher baseline levels of CGRP compared to healthy controls. Furthermore, patients in active phase (p < 0.001) and in remission (p < 0.001) had higher CGRP levels compared to migraine patients. We found no differences in PACAP38 and VIP levels between CH patients, migraine patients and healthy controls.
In patients in an active disease state, we found no difference in baseline CGRP, PACAP38 or VIP levels in patients on prophylactic treatment versus those without (p > 0.05). There was no correlation between hours since last attack, nor 30-day attack burden and baseline level of CGRP, PACAP38 or VIP (p > 0.05).
Discussion
The main finding of the present study was that CH patients in remission had higher baseline levels of CGRP, but not VIP or PACAP38, compared to chronic CH patients. Furthermore, CGRP-induced CH attacks were not associated with elevated levels of CGRP, VIP or PACAP38. In addition, CH patients, irrespective of disease phase, had higher plasma levels of CGRP compared to migraine patients and healthy controls.
A novel finding of the present study was that chronic CH patients had lower plasma CGRP compared to CH patients in remission. This suggests that plasma levels of CGRP may fluctuate with disease activity. The results are very interesting when viewed in the light of recent press releases on the preventive effect of the anti-CGRP monoclonal antibodies in episodic patients in active phase, but not in patients with chronic CH. Interestingly, poor treatment response in chronic CH is reported across different treatment modalities, indicating basic pathophysiological differences between phenotypes (19,20). The question is, why did chronic CH patients have lower CGRP levels than CH patients in remission and episodic CH patients in active phase? Precisely how the regulation of CGRP expression and release aligns with with our data is difficult to reconcile, but CGRP expression has been reported to be regulated in many different ways. For example, CGRP levels are elevated in animal models of nerve damage, nerve regeneration and tissues undergoing inflammatory response (21). CGRP is expressed and released from C fibers and its receptors are present in A delta fibers (22). In man, application of capsaicin to the nasal mucosa in one study led to immediate release of CGRP in saliva and plasma (8). It is possible that secreted CGRP may act in an autocrine fashion to further increase CGRP release in a positive feedback loop, a mechanism possibly implicated in peripheral sensitization (23). In rats, a single subcutaneous capsaicin injection in the hind-paw depleted CGRP levels in the skin and sciatic nerve after 8 and 10 days (24,25). Interestingly, repeated capsaicin applications to the nasal mucosa resulted in desensitization and time-dependent recovery of responses (26). In addition, intradermal capsaicin injections in another study produced a steady increase of CGRP levels in the first sampling period but failed to reach significance in the second session (27). Thus, capsaicin-induced desensitization of sensory afferents might lead to depletion of neuropeptide release from afferents or decreased activity of transient receptor potential vanilloid 1 channels (28,29). Taken together, these data suggest that chronic CH patients may exhibit low plasma CGRP due to depletion of CGRP from trigeminal afferents. Whether a CH attack represents a comparable stimulus to capsaicin is unknown, but it is possible that endogenous processes influence CGRP expression. Several factors might have influenced our data. We tested for possible influence of a recent attack and attack burden (frequency) and found that CGRP levels were not associated to the most recent attack or to attack burden in the 30 days preceding baseline sampling. Exogenous factors such as use of preventive and abortive treatments may theoretically influence CGRP expression. One study reported that treatment with corticosteroids can reduce CGRP levels in episodic CH patients (4). In the present study, 61.5% of chronic CH patients took preventive treatments compared to 44.4 % of episodic CH patients, but an exploratory analysis revealed no difference in baseline levels. Preclinical studies reported that application of 5-HT1 receptor agonist decreased the synthesis of CGRP in the trigeminal ganglion (TG) (30) and that 7-day infusion of sumatriptan upregulated CGRP expression in trigeminal dural afferents (31). In the present study, we did not record the patients' use of triptans prior to baseline sampling, but in future studies investigating CGRP in CH, this should be taken into consideration.
In the post hoc analyses we found that episodic patients in active phase and in remission had higher baseline levels of CGRP than episodic migraine patients and healthy controls. Although data from migraine patients and healthy controls is historical, and thus should be interpreted with caution, it is an interesting observation. One study reported that chronic migraine patients had higher CGRP levels than episodic migraine patients, healthy controls or episodic CH patients in remission (32). This study also reported no difference between patients with episodic migraine and CH patients (32). As different assays have been used across studies in migraine and CH it is impossible to compare results directly, and further hypothesis-based studies are necessary to address possible difference in CGRP levels between CH and migraine.
Collectively, our data suggest that CGRP may be altered in CH, but the findings should be reproduced in a larger cohort of CH patients, ideally in prospective studies investigating changes in CGRP over time and disease state.
In the present study, chronic CH patients had lower baseline levels of PACAP38 compared to episodic patients in active phase, but similar levels compared to CH patients in remission. In line with our CGRP findings in chronic CH patients, this suggests possible pathophysiological differences between disease states. Another important finding was that CGRP-induced CH attacks were not associated with alterations in plasma PACAP38 or VIP. These data are in contrast two previous studies, where VIP (7) and PACAP38 (9) were reported to be elevated during spontaneous attacks. In the current study, chronic patients had an average longer disease duration compared to episodic patients, which might be an influencing factor. A migraine study reported that baseline levels of PACAP38 were negatively correlated with disease duration (33), suggesting that longer disease duration or transition to the chronic phase alters the regulation of this peptide. In support, repeated chemical stimulation of dura surrounding the superior sagittal sinus decreased PACAP38 levels in the TG in rats (34). Both PACAP38 and VIP may be considered as markers of parasympathetic activation. Higher PACAP38 levels in episodic CH patients in active phase compared to chronic patients could theoretically reflect marked parasympathetic activation, but as VIP was unchanged, this interpretation remains speculative. Of note, we found no correlation between hours since last attack and baseline VIP or PACAP38 levels, as would be expected given the short half-lives (min) of VIP and PACAP38 (35). Infusion of CGRP induced an increase in VIP, but not PACAP38, and development of an attack was associated with a decrease in VIP. As VIP levels were highest immediately following infusion and attacks occurred on average 34 min after onset of infusion, the observed attack-associated decrease in VIP was likely to coincide with the natural fall in VIP. Interestingly, our previous study using the same assay also found elevated VIP after CGRP infusion in migraine patients (12). These findings suggest that CGRP infusion is associated with transient elevation of plasma VIP, but it does not seem to be associated with attack development. Collectively, CH disease activity was not associated with elevation of PACAP38 and VIP. Furthermore, CGRP-induced CH attack did not increase plasma VIP and PACAP38, which is in contrast to previous studies (7,9). The inconsistency of results across studies is likely attributed to use of different assays (35).
We collected blood from the antecubital vein, and not from the external jugular vein. One might argue that plasma levels should be collected in the cranial outflow. In migraine patients, elevated plasma CGRP was reported to be elevated in peripheral blood ictally (36) and interictally (37). One study in migraine patients reported no changes in plasma CGRP during attacks. To date, no studies have compared plasma PACAP38 and VIP between the two sampling sites, but elevated levels of these neuropeptides were reported in peripheral blood during and outside of migraine attacks (9,33). We acknowledge relatively small sample size in the present study. However, we obtained two sets of samples for baseline comparisons (in total 62 samples) and performed a robust statistical analysis. We collected “attack samples” in 11 patients. Previous studies reported plasma alterations during provoked and spontaneous attacks in an avarage of 14 patients (5–7,9). Thus, the present sample size should be sufficient to detect possible attack-related changes in neuropeptides. As blood samples were not drawn in all provoked attacks, an element of selection bias might affect attack results. However, as these results were negative, the concern for this bias seems less important. With regards to sensitivity and specificity of CGRP assay used in the current study, we measured a robust elevation of CGRP after CGRP infusion and importantly no concomitant increase in PACAP38, which is structurally similar.
The present study demonstrated that CH disease activity might be associated with alterations of CGRP expression, possibly PACAP38, but not VIP expression. Future studies should investigate the potential of using CGRP measurements in monitoring of disease state and predicting response to preventive treatments, including response to anti-CGRP monoclonal antibodies.
Footnotes
Article highlights
The present study demonstrated that cluster headache disease activity is associated with alterations of CGRP expression, possibly PACAP38, but not VIP expression.
Our results indicate that there are basic pathophysiological differences between episodic and chronic cluster headache patients.
The observed lower CGRP levels in chronic cluster headache patients at baseline might offer an explanation as to why anti-CGRP monoclonal antibodies have proven effective in episodic but not in chronic cluster headache patients.
Acknowledgements
The authors would like to thank all participating patients in the study. Without their willingness to subject themselves to a potential CH attack, this study would not have been possible. The authors would furthermore like to thank study nurses Mette Frank Fisker and Mette Bisgaard for helping with recruitment of patients and handling of blood samples on study days.
Author contributions
Agneta Snoer: Study concept and design, acquisition of data, analysis (including statistical analyses) and interpretation, drafting the manuscript. Anne Luise Haulund Vollesen: Study concept and design, acquisition of data, analysis and interpretation, drafting the manuscript. Rasmus Beske: Acquisition of data and critical revision of the manuscript for important intellectual content. Song Guo: Study concept and design, critical revision of the manuscript for important intellectual content. Jan Hoffmann: Study concept and design, critical revision of the manuscript for important intellectual content. Jan Fahrenkrug: Analyses of biochemical data, interpretation and critical revision of the manuscript for important intellectual content. Niklas Rye Jørgensen: Analyses of biochemical data, interpretation and critical revision of the manuscript for important intellectual content. Torben Martinussen: Statistical analyses and critical revision of the manuscript for important intellectual content. Rigmor Jensen: Study concept and design, interpretation of study result, supervision. Messoud Ashina: Study concept and design, interpretation of study result, critical revision of the manuscript for important intellectual content and supervision.
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship and/or publication of this article: Agneta Snoer is a sub-investigator on clinical trials sponsored by Eli Lilly; Anne Luise Haulund Vollesen has received personal fees from Teva Pharmaceutical Industries; Rasmus Beske and Song Guo report no competing interests. Jan Hoffmann has consulted for and/or serves on advisory boards for Allergan, Autonomic Technologies Inc (ATI), Chordate Medical AB, Eli Lilly, Hormosan Pharma, Novartis and Teva Pharmaceutical Industries and received honoraria for speaking from Allergan, Autonomic Technologies Inc. (ATI), Chordate Medical AB, Novartis and Teva Pharmaceutical Industries. Jan Fahrenkrug, Niklas Rye Jørgensen and Torben Martinussen report no competing interests. Rigmor Højland Jensen has given lectures for Pfizer, Berlin-Chemie, Norspan, Merck and Autonomic Technologies (ATI), and a principal investigator on clinical trials sponsored by Eli Lilly and Autonomic Technologies Inc. (ATI). Messoud Ashina is a consultant, speaker, or scientific advisor for Allergan, Amgen Inc, Alder BioPharmaceuticals, ATI Technologies, Eli Lilly and Company, Novartis, and Teva Pharmaceutical Industries and is primary investigator for Amgen 20120178 (phase 2), 20120295 (phase 2), 20130255 (OLE), 20120297 (phase 3), Alder ALD403-CLIN-001 (phase 3), Amgen PAC1 20150308 (phase 2a), and GM-11 gamma-Core-R trials.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The work has been supported by grants from The Lundbeck Foundation (grant R155-2014-171) Novo Nordisk Foundation (grant NNF11OC101433), Tryg Foundation and Research Foundation of Rigshospitalet.
*These authors contributed equally to this work