
Abstract
Background
Stress is the most commonly reported migraine trigger. Dynorphin, an endogenous opioid peptide acting preferentially at kappa opioid receptors (KORs), is a key mediator of stress responses. The aim of this study was to use an injury-free rat model of functional cephalic pain with features of migraine and medication overuse headache (MOH) to test the possible preventive benefit of KOR blockade on stress-induced cephalic pain.
Methods
Following sumatriptan priming to model MOH, rats were hyper-responsive to environmental stress, demonstrating delayed cephalic and extracephalic allodynia and increased levels of CGRP in the jugular blood, consistent with commonly observed clinical outcomes during migraine. Nor-binaltorphimine (nor-BNI), a long-acting KOR antagonist or CYM51317, a novel short-acting KOR antagonist, were given systemically either during sumatriptan priming or immediately before environmental stress challenge. The effects of KOR blockade in the amygdala on stress-induced allodynia was determined by administration of nor-BNI into the right or left central nucleus of the amygdala (CeA).
Results
KOR blockade prevented both stress-induced allodynia and increased plasma CGRP. Stress increased dynorphin content and phosphorylated KOR in both the left and right CeA in sumatriptan-primed rats. However, KOR blockade only in the right CeA prevented stress-induced cephalic allodynia as well as extracephalic allodynia, measured in either the right or left hindpaws. U69,593, a KOR agonist, given into the right, but not the left, CeA, produced allodynia selectively in sumatriptan-primed rats. Both stress and U69,593-induced allodynia were prevented by right CeA U0126, a mitogen-activated protein kinase inhibitor, presumably acting downstream of KOR.
Conclusions
Our data reveal a novel lateralized KOR circuit that mediated stress-induced cutaneous allodynia and increased plasma CGRP in an injury-free model of functional cephalic pain with features of migraine and medication overuse headache. Selective, small molecule, orally available, and reversible KOR antagonists are currently in development and may represent a novel class of preventive therapeutics for migraine.
Abbreviations
KOR = kappa opioid receptor; MOH = medication overuse headache; CeA = central nucleus of amygdala; BLA = basolateral amygdala; CGRP = calcitonin gene-related peptide; MEK = mitogen-activated protein kinase kinase; PAG = periaqueductal gray; CRF = corticotrophin releasing factor; HPA = hypothalamus-pituitary-adrenal; AOC = area over the time effect curve.
Introduction
Migraine is the most prevalent neurological disorder, the seventh leading cause of disability worldwide, and presents an enormous global burden (1). Approximately 12% of the general population suffers from migraine and about a third of these patients have more than three migraine attacks per month debilitating enough to require bed rest (2). Chronic migraine, defined as the presence of headache on more than 15 days per month with at least eight days per month meeting the criteria for migraine with or without aura, is a severe and disabling form of migraine that affects approximately 1–2% of the population. Frequency of headache and the overuse of acute medications (defined as use on more than 10 days per month for most medications) are the major risk factors that lead to the progression from episodic to chronic migraine and medication overuse headache (MOH) (3). MOH is the 20th leading cause of Years Lived in Disability (YLD), and posts an enormous burden to society (1). The majority of individuals with migraine utilize medications for acute treatment. However, many individuals are not satisfied with their acute treatment; many do not tolerate, respond to, or are able to use migraine-specific acute medications because of contraindications. Individuals with frequent migraine or chronic migraine are candidates for preventive treatments (4). Preventive therapies include drugs from different mechanistic classes such as tricyclic antidepressants (5,6), anticonvulsants (e.g. topiramate) (7,8), β-blockers (e.g. propranolol) (9), calcium channel blockers (10,11) and onabotulinum toxin A (12). In many patients, these drugs are associated with limited efficacy and numerous undesirable side effects (for review, see (4)). Monoclonal antibodies (mAbs) targeting calcitonin gene-related peptide (CGRP), or its receptor, have been shown to be clinically effective as migraine preventive agents in phase 2 clinical trials (13–15). However, monoclonal antibodies have a terminal half-life of three to six weeks or longer, and conclusions about the long-term safety associated with persistent blockade of CGRP for long periods of time, especially in patient populations with specific vascular risk factors, must await long-term safety studies and a large sample of patient exposures (16). New efficacious therapies that are orally available, reversible, and safe may fill clinical needs and allow optimization of therapy for patients.
Stress is consistently identified as the most common migraine trigger (17). The relationship of stress to migraine is complex, with observations that the stress itself, or commonly the relief of stress, may elicit a migraine attack (18). How stress may trigger migraine is not well understood. Acute stress is adaptive and necessary for survival, inducing the sympathetic “fight or flight” response. However, repeated, chronic, or uncontrolled stress is maladaptive and induces “allostatic overload” associated with changes in brain circuits (19). The dynorphin/kappa opioid receptor (KOR) system has recently emerged as a novel therapeutic target for the treatment of stress-related mood disorders including anxiety, depression, drug seeking and relapse (20–25). The KOR is widely expressed in structures involved in the modulation of reward, mood state, and cognitive function including the ventral tegmental area (VTA), nucleus accumbens (NAc), prefrontal cortex, hippocampus, striatum, amygdala, locus coeruleus, substantia nigra, dorsal raphe nucleus, and hypothalamus in both the rodent and human brain (22,24,26–28). Stress-induced activation of the hypothalamus, in part through facilitatory reciprocal connections with the amygdala (29), can elicit release of corticotropin releasing factor (CRF) and activation of CRF1 receptors in several brain regions, including the amygdala, VTA and neocortical sites (25,30). Activation of these sites by behavioral stress or exogenous CRF causes dynorphin release and phosphorylation of the KOR in the amygdala, VTA, dorsal raphe nucleus and NAc (21,22). The stress response has been suggested to involve a hypothesized CRF to dynorphin to KOR pathway (31). Importantly, activation of the hypothalamus has been repeatedly implicated in the earliest phase (premonitory) of a migraine attack (32–34). These observations lead us to hypothesize that KOR antagonists may serve as effective preventive therapies for stress-related migraine.
We have previously described an “injury-free” model of cephalic pain based on the clinical observation of increased frequency of migraine attack following overuse of acute medications, including triptans or opioids, a condition termed MOH (35). In this model, priming animals with sumatriptan for one week results in a state of “latent sensitization” characterized by long lasting effects including (a) increased numbers of trigeminal ganglion cells back-labeled from the dura mater that express CGRP; (b) increased sensitivity to putative migraine triggers including nitric oxide (NO) donors or environmental stress; (c) delayed and generalized stress-induced cutaneous allodynia and (d) increased levels of CGRP in the jugular blood (36–38). We have recently shown that stress-induced cephalic and extracephalic allodynia in rats with prior exposure to sumatriptan is blocked by the CGRP antibody TEV48125, increasing the relevance of this model (36).
The aim of this study was to use this injury-free model relevant to MOH to test the possible preventive benefit of KOR blockade on stress-induced cephalic pain. Here, we treated rats with sumatriptan for seven days followed by a 14-day drug free period; then, on two consecutive days, we exposed them for one hour without restraint to bright light as an environmental stress (BLS). We assessed outcome measures that are likely to have translational value, including cephalic and extracephalic (i.e. hindpaw) allodynia and levels of CGRP from the jugular blood, commonly observed during migraine attacks (39,40), in the presence or absence of systemic or intra-amygdala blockade of KOR signaling. Our results indicate that in “primed” animals, stress-induced allodynia is mediated by the dynorphin/KOR signaling in the CeA, and that blockade of this system may represent a promising strategy for migraine prevention.
Materials and methods
Experimental design
The aim of these studies was to evaluate the effect of KOR antagonist in stress-induced outcome measures with putative relevance to MOH and migraine (experimental time line in Supplementary Figure S1a). The studies were performed over a period of 20 months using behavioral (cutaneous cephalic and extracephalic allodynia, tail-flick in mice), neurochemical (plasma CGRP release, dynorphin, KOR phosphorylation) and immunohistochemical (KOR phosphorylation,) assessment of environmental stress following priming of rats with a transient period of sumatriptan treatment. Different cohorts of animals were used to assess approximately 57 experimental conditions, averaging approximately nine animals per group. All experiments followed the ARRIVE guidelines wherever possible. Following randomization of animals, behavioral testing was performed by experimenters blinded to treatment conditions by coding solutions prior to injection, and by having different individuals perform group and dose pairings. Blinding codes were not revealed until the conclusion of the experiment. Fourteen days following priming with either sumatriptan or saline, periorbital and hindpaw thresholds were measured in the same rats. Rats that responded to von Frey stimulation with thresholds lower than 7 grams or 12 grams for cephalic or hindpaw thresholds were excluded from the study. In cases where an animal passed one of the sensory threshold measures, it was included in the study for that endpoint in order to minimize animal numbers, sometimes resulting in unequal group sizes. Overall, approximately 10–15% of rats were discarded due to a disqualified periorbital or hindpaw baseline and there being no differences between the groups previously primed with sumatriptan or with saline. Mice were used only to characterize the time course of KOR antagonist blockade to minimize the numbers of rats used.
Animals
Adult male Sprague-Dawley rats (starting weight 175–200 g, Harlan, Indianapolis, IN, USA) or male C57BL/6J mice (weight 20–25 g, Harlan, Indianapolis, IN, USA, for tail flick study only) were used. All procedures were performed in accordance with the guidelines of the Committee for Research and Ethical Issues of IASP under protocols approved by the University of Arizona Institutional Animal Care and Use Committee. Animals were initially housed three per cage for rats and five per cage for mice on a 12 h light-dark cycle with food and water ad libitum. If animals received brain cannulation, they were then individually housed.
Implantation of minipumps
Surgical implantation (s.c.) of osmostic minipumps (model 2001; Alzet) was performed as previously described (38). Minipumps were loaded with sumatriptan (0.6 mg/kg/day) or vehicle (0.9% saline), which allowed for continuous release at a rate of 1 µl/h for seven days. Gentamicin (8 mg/kg, s.c.) was given following surgery to prevent infection.
Intracranial cannulation
Stereotaxic surgeries were performed at day 14 post-mini-pump implantation (as described previously (41). Brain loci coordinates were obtained and verified according to the Paxinos and Watson Rat Brain Atlas (42). A single guide cannula (Plastics One, Roanoke, VA, USA) was implanted into the right or left CeA (AP: bregma −2.0 mm; ML: midline ± 4.0 mm; DV: skull −7.0 mm). Rats were housed individually and allowed to recover for at least six days. Solutions (0.5 µl) were administered slowly through injectors that extended 1 mm beyond the guide cannula.
Evaluation of periorbital and hindpaw cutaneous allodynia
Cephalic and extracephalic cutaneous allodynia was evaluated as described previously (43) with calibrated von Frey filaments (Stoelting, Wood Dale, IL, USA). The filaments were applied perpendicularly to the periorbital region at the center of the forehead or the plantar surface of the hindpaws until a withdrawal response was elicited. Cutoff was set at 8 or 15 g for periorbital or hindpaw regions, respectively. The tactile threshold from the left hindpaw was evaluated unless specifically noted. Withdrawal thresholds were determined by Dixon nonparametric test and expressed as the mean withdrawal threshold (44,45).
Tail flick test
Mice were held gently and 1/2 to 2/3 of the tail was dipped into 50℃ hot water. The latency to a rapid flick of the tail was recorded. The cut-off time was set at 15 s to prevent tissue damage.
Sumatriptan priming and environmental stress
Rats were treated with sumatriptan (0.6 mg/kg/day) or saline (s.c.) delivered by osmotic minipumps along with either a single injection of nor-BNI (3 mg/kg/injection, s.c.) at day 0 or with three injections at days 0, 2, and 4, followed by a 14-day drug free period. Exposure to BLS was performed on days 20 and 21 post-sumatriptan treatment (46). Following the second BLS episode, periorbital and hindpaw withdrawal thresholds were tested at 1 h intervals over a 5 h time course. Separate cohorts of animals were used to evaluate systemic or unilateral intra-amygdala administration of a KOR agonist, U69,593, or of the short or long-acting KOR antagonists, CYM51317 and nor-BNI, respectively, or of the MEK inhibitor U0126 against BLS-induced allodynia. Intracranial injections were verified histologically following all testing, and data from animals with incorrect cannula placement or clogged cannulas were excluded from data analysis (<10%). Each experiment was repeated internally at least twice. Outliers were identified using Graphpad Prism 6.0 Grubbs’ test (maximum one outlier per group allowed) and removed from the data analysis.
Rats were exposed to bright light for one hour on Day 20 and Day 21 (D20 and D21) as described previously (46). Two halogen lights were placed 350 cm from both of the larger sides of the clear Plexiglas cage to deliver bright light (intensity between 3200 lux to 3500 lux measured at the center of the cage floor). With these parameters, the temperature change inside the cage varies by less than 1℃. The animals were allowed to acclimate to a new cage and exposed to bright light for two consecutive days for 1 h. Animals were then returned to their home cages afterwards. At day 21, animals were acclimated to von Frey chambers and the tactile threshold was measured for periorbital and hindpaw regions before the second exposure of bright light, and again each hour following the second bright light exposure until sensory thresholds normalized.
CGRP plasma levels
CGRP levels in plasma were measured with ELISA kits (Cayman Chemical, Ann Harbor, MI, USA). Approximately 2 ml of blood was collected from the jugular vein under isoflurane (2%) anesthesia and placed into EDTA-coated Vacutainers (Becton Dickenson, Franklin Lakes, NJ, USA). Blood samples were centrifuged at 3000 rpm at 4℃ for 20 min and supernatant (plasma) was extracted and stored at −80℃. CGRP concentrations in each sample were measured as duplicates via ELISA and the average of the duplicates was calculated to represent the sample concentration.
Amygdala dynorphin content
Rats were decapitated following isoflurane (2%) anesthesia at 2 h post second stress. A 1 mm punch with 1 mm thickness was collected from the left and right CeA and immediately frozen using liquid nitrogen and stored at −80℃ for analysis. Dynorphin was extracted from the amygdala tissues following the methods provided with the dynorphin ELISA kits (Phoenix Pharmaceuticals, CA, USA). The dynorphin concentrations were measured in duplicates and the average of the duplicates was calculated to represent the sample concentration.
Phosphorylated KOR immunofluorescent detection
Rats were anesthetized 2 h after BLS by i.p. ketamine/xylazine and transcardially perfused with 4% paraformaldehyde. Coronal brain sections (20 µm thick) were cut in a Microm HM 525 cryostat and mounted on Superfrost Plus microscope slides. Brain sections were permeabilized with 0.2% TritonX100, blocked with 1% BSA and 5% normal goat serum and incubated overnight with rabbit polyclonal anti-phospho-Ser369 KOR (1:1,000; LifeSpan BioSciences, Inc.). The sections were incubated with biotinylated anti-rabbit antibody followed by the ABC complex (Vectastain Elite ABC kit, Vector Labs) and Tyramide Signal Amplification detection (TSA Plus Fluorescein Kit, Perkin Elmer, Waltham, MA, USA). Slides were mounted in Vectashield mounting medium (Vector Lab. Inc., Burlingame, CA, USA) and examined under an Olympus BX51 microscope equipped with a Hamamatsu C8484 digital camera. Micrographs of CeA and BLA of 2–6 sections per rat 100 µm apart within the Bregma −2.0 to −2.7 mm were taken, and the amount of pKOR immunofluorescence in the CeA was analyzed by the ImageJ software. Phosphorylated KOR immunofluorescence in the CeA was normalized to the intensity of the adjacent baselateral amygdala (BLA) to minimize the variations among slides. The ratio between CeA and BLA mean intensity was calculated as a measure of a relative increase of pKOR in the CeA.
Drugs
Sumatriptan (Abmole Bioscience, Houston, TX, USA), nor-BNI (long-acting KOR antagonist, Tocris, Bristol, BS, UK), and U0126 (MEK inhibitor, Tocris, Bristol, BS, UK) were dissolved in saline (0.9% NaCl). CYM51317 (short-acting KOR antagonist, synthesized by Dr Ed Roberts, Scripps, Supplementary Figure 1) was dissolved in 10% DMSO/10% Tween 80 and 80% DiH2O. U69,593 (KOR agonist, Tocris, Bristol, BS, UK) was dissolved in a small amount of HCl first, then diluted with normal saline followed by the final adjustment of pH to neutral using NaOH. U50488 (KOR agonist, Tocris, Bristol, BS, UK) was dissolved in saline. The dose volume was 0.5 µl/rat for CeA, 1 ml/kg for s.c. or i.p. injections to rats, and 10 ml/kg for s.c. or i.p. injections in mice.
Statistical analysis
All results are expressed as mean ± SEM. Statistical analyses were calculated using GraphPad Prism 6 software. Due to the multiple outcome measures and exploratory feature of this study, a priori power calculations were not performed. In cases where treatments were found to be ineffective, post-hoc power analysis was performed to ensure adequate power in the conclusions. The area over the time effect curve (AOC) was calculated with JFlashcalc software developed by Dr Michael Ossipov (www.u.arizona.edu/∼michaelo). The unpaired t-test (two-tailed) was used when only two groups were compared. For three or more group comparisons, one-way ANOVA followed by Fisher’s Least Significant Difference (LSD) test or two-way ANOVA post-hoc Dunnett or Sidak’s multiple comparisons test was applied. The F(DFn,DFd) and p values are reported for the ANOVA tests in Supplementary Table 1; post-hoc comparison is reported as “significant” or not significant without the actual p value. Significance was set at p < 0.05.
Results
CYM51317 is a novel short-acting KOR antagonist
The duration of action of the KOR antagonists nor-BNI and CYM51317, a recently synthesized KOR antagonist (47) (Supplementary Figure S2), were determined in the mouse hot water (50℃) tail flick test by their ability to antagonize the antinociceptive effects of U69,593, a selective KOR agonist. KOR antagonists or vehicle were administered at 1 h, 24 h, or 7 d prior to U69,593 (10 mg/kg, i.p.), and latencies for tail-withdrawal measured at baseline and 30 minutes after agonist injection (Supplementary Figure S3). Baseline tail-flick latency of naïve mice averaged 8.6 ± 0.3 s. Injection of U69,593 produced antinociception indicated by a significant increase in tail-flick latency. Pretreatment with nor-BNI (3 mg/kg, s.c.) at 1 or 24 hr, but not seven days prior to U69,593, blocked antinociception. In contrast, CYM51317 (20 mg/kg, i.p.) blocked the U69,593-induced antinociception only at the 1 hr pretreatment time point, confirming a short duration of action in blocking KOR.
Systemic KOR antagonist nor-BNI prevented stress-induced cephalic and extracephalic allodynia
The possible effects of systemic nor-BNI on stress-induced allodynia were evaluated using two different protocols. In the first protocol, nor-BNI (3 mg/kg, s.c.) or vehicle was injected on days 0, 2 and 4 during osmotic minipump infusion of sumatriptan to provide sustained blockade of KOR signaling. Two weeks after the termination of the sumatriptan infusion, the rats were exposed on two consecutive days to bright light stress (BLS) and periorbital and hindpaw allodynia were recorded (Figure 1 (a) and (b)). Sumatriptan exposure with s.c. vehicle injections followed by BLS resulted in a significant reduction in periorbital thresholds, indicative of cutaneous allodynia, at 2 and 3 h after BLS. Similar observations were made with hindpaw allodynia. In contrast, repeated injections of nor-BNI at days 0, 2 and 4 during the sumatriptan priming period blocked the development of stress-induced periorbital and hindpaw allodynia. In the second protocol, nor-BNI (3 mg/kg, s.c.) was given as a single injection on day 0 of sumatriptan infusion. Tactile thresholds were significantly reduced by BLS to a lesser extent than observed with repeated dosing (Figure 1 (a) and (b)). The integrated area over the time-effect curve (AOC) for periorbital and hindpaw withdrawal thresholds also demonstrated that repeated injections of nor-BNI during the sumatriptan infusion fully blocked the AOC after BLS, while a single injection of nor-BNI on day 0 produced a partial, but significant, block of BLS-induced cutaneous allodynia (Figure 1 (c) and (d)).
Systemic Nor-BNI, a long-acting KOR antagonist, blocks bright light stress (BLS)-induced periorbital or hindpaw allodynia and CGRP release. (a), (b): Nor-BNI (3 mg/kg, s.c.) was injected at two day intervals (days 0, 2, 4) during infusion with sumatriptan (0.6 mg/kg/day for seven days, s.c. with osmotic minipump) or as a single s.c. injection (3 mg/kg, s.c.) at day 0 of the sumatriptan administration; saline was infused by minipump and given as a single injection (vehicle of nor-BNI) in control groups. Periorbital (a) and hindpaw (b) allodynia was evaluated 21 days after the initiation of sumatriptan infusion following exposure to BLS. Repeated nor-BNI administration during sumatriptan infusion significantly blocked periorbital and hindpaw allodynia. A single nor-BNI injection at minipump implantation (D0 only) partially, but significantly blocked BLS-induced periorbital and hindpaw allodynia. *p < 0.05, **p < 0.01, ***p < 0.001 vs. baseline (BL). Group sizes for each treatment are shown on the figure. (c), (d): Group comparisons of Area Over the Curve (AOC) for periorbital (c) and hindpaw (d) allodynia demonstrate that repeated administration of nor-BNI (D0, 2, 4) during sumatriptan infusion significantly blocked BLS-induced cutaneous allodynia. A single administration of nor-BNI at the time of minipump implantation (D0 only) partially, but significantly attenuated tactile hypersensitivity. *p < 0.05, **p < 0.01, ***p < 0.001 vs. vehicle control; ## p < 0.01, ### p < 0.001 vs. saline-primed animals. (e): BLS significantly elevated CGRP levels in the plasma at 30 min, but not 2 h post-BLS in sumatriptan primed rats (N = 13 and 9 for saline-primed rats at 30 min and 2 h, respectively; and 8 and 11 for sumatriptan-primed rats at 30 min and 2 h, respectively; *p < 0.05). (f): Treatment with nor-BNI during sumatriptan infusion (D0, 2, 4) blocked BLS-induced plasma CGRP elevation at 30 min post-BLS (N = 9 and 9 for saline infusion plus vehicle and saline plus nor-BNI, respectively; and 8 and 5 for sumatripan plus vehicle and sumatriptan plus nor-BNI, respectively; **p < 0.01, ***p < 0.001). Data represent mean ± SEM. Statistical analysis was performed using two-way ANOVA followed by Dunnett’s ((a) and (b)) or Sidak’s ((e) and (f)) multiple comparisons post-hoc test; and one-way ANOVA followed by Fisher’s LSD post-hoc test for (c) and (d).
Systemic KOR antagonist nor-BNI blocked stress-induced increase in plasma CGRP
We have previously shown that BLS increased plasma, but not CSF, CGRP levels in sumatriptan-primed rats (36). We examined CGRP levels in blood samples taken from the jugular vein at 30 min and 2 h after the second BLS episode and confirmed that BLS significantly elevated plasma CGRP levels in sumatriptan-primed rats when compared to the saline-primed group at the 30 min, but not 2 h timepoint (Figure 1 (e)). Based on this observation, we collected plasma at the 30 min time point for the nor-BNI study. Administration of nor-BNI (3 mg/kg, s.c.) on days 0, 2 and 4 during the sumatriptan infusion prevented the BLS-induced increase in plasma CGRP levels (Figure 1 (f)). There were no significant differences in CGRP levels between vehicle and nor-BNI treatment groups in saline-primed rats after stress.
Oral CYM51317 administration before BLS prevented cephalic and extracephalic cutaneous allodynia
We used the short-acting KOR antagonist, CYM51317, to determine whether blocking KOR signaling prior to and during BLS is sufficient to prevent the expression of stress-induced allodynia (Figure 2). Oral administration of CYM51317 (20 mg/kg), given 30 min before each BLS exposure, prevented the expression of periorbital and hindpaw allodynia evaluated following the second BLS in sumatriptan-primed rats (Figure 2 (a) and (b)). Saline-infused animals injected with vehicle or CYM51317 did not develop cutaneous allodynia. The AOC data from periorbital and hindpaw threshold (Figure 2 (c) and (d)) also demonstrated that CYM51317 (20 mg/kg, p.o.) administration abolished stress-induced cutaneous allodynia in sumatriptan-primed rats.
CYM51317, a short-acting KOR antagonist, blocks BLS-induced periorbital or hindpaw cutaneous allodynia. (a), (b): BLS-induced periorbital (a) and hindpaw (b) allodynia in sumatriptan pre-exposed rats was blocked by systemic administration of CYM51317 (20 mg/kg, p.o.) at 1 h prior to each BLS episode. ***p < 0.001 vs. baseline (BL). Group sizes are shown on the figure. (c), (d): Group comparisons of Area Over the Curve (AOC) for periorbital (c) and hindpaw (d) allodynia demonstrate that administration of CYM51317 (20 mg/kg, p.o.) prior to each BLS episode significantly blocked BLS-induced tactile hypersensitivity. **p < 0.01, ***p < 0.001. Data represent mean ± SEM. Statistics performed using two-way ANOVA with Dunnett’s multiple comparisons post-hoc test for (a) and (b); and one-way ANOVA followed by Fisher’s LSD post-hoc test for (c) and (d).
BLS increases dynorphin content and pKOR expression in the central nucleus of amygdala (CeA)
We evaluated whether sumatriptan “priming” would result in enhanced dynorphin/KOR activity in the CeA in response to stress. We measured dynorphin content in the CeA using ELISA as well as phosphorylation of the KOR at Ser 369 (pKOR) as a measure of functional consequences of agonist activation of the receptor using immunohistochemistry (Figure 3).
Dynorphin content and KOR activation in the central nucleus of the amygdala (CeA). (a), (b): BLS elevated dynorphin levels in the right (a) and left (b) CeA of sumatriptan, but not saline, pre-exposed rats at 2 h (N = 15 and 19 for saline primed rats without and with BLS; and 17 and 19 for sumatriptan primed rats without and with BLS; *p < 0.05, **p < 0.01 vs. Saline plus BLS). (c): A representative coronal section of the brain of a sumatriptan-primed rat exposed to BLS (at approximately Bregma −2.5 mm). The amount of pKOR immunofluorescence in the CeA was analyzed by the ImageJ software and normalized to the intensity of the adjacent basolateral amygdala (BLA). The ratio between CeA and BLA mean intensity was calculated as a measure of a relative increase of pKOR in the CeA. Scale bar = 100 µm. (d): Relative CeA pKOR intensity on both sides was increased in the sumatriptan plus BLS group at 2 h post-stress (vs. sumatriptan BL or saline plus BLS, ***p < 0.001; N = 3 and 4 for saline baseline and saline plus BLS; and 4 and 7 for sumatriptan baseline and sumatriptan plus BLS). Data are reported as the mean ± SEM. Statistics performed using two-way ANOVA followed by Dunnett’s ((a) and (b)) or one-way ANOVA followed by Fisher’s LSD post-hoc test for (d).
Saline-primed and sumatriptan-primed rats showed no significant differences in right and left CeA dynorphin levels prior to BLS exposure (Figure 3 (a) and (b)). However, BLS exposure resulted in a significant elevation of dynorphin levels at the 2 h time point in the right and left CeA of sumatriptan-primed rats relative to those of saline-primed rats.
To evaluate the anatomical location of dynorphin/KOR activity, sumatriptan or saline-primed animals were perfused 2 h after the second BLS and coronal brain sections containing the amygdala were immunolabeled with pKOR and examined using fluorescent microscopy. The mean pKOR fluorescent intensity of a spherical region of interest (ROI) in the CeA was quantified using ImageJ and normalized to the mean fluorescent intensity of the same spherical ROI in the BLA (Figure 3 (c), (d), S5). The relative CeA/BLA pKOR did not differ between saline and sumatriptan- treated rats prior to stress. Following BLS, however, the relative CeA/BLA pKOR intensity increased significantly on both right and left sides in sumatriptan, but not saline, pre-treated rats.
Collectively, these data demonstrate that sumatriptan priming or BLS alone does not result in activation of the dynorphin/KOR system in the amygdala. However, in sumatriptan-primed rats, BLS increases dynorphin levels and results in activation and phosphorylation of the KOR receptors in both the left and right CeA.
Nor-BNI administration into the right, but not left, CeA blocked BLS-induced allodynia
We determined whether microinjection of nor-BNI into the right or left CeA would prevent BLS-evoked allodynia in sumatriptan-primed rats. Nor-BNI (2.5 µg) was microinjected 1 h prior to the first BLS exposure and cutaneous allodynia was measured following the second exposure to BLS on the following day (Figure 4). Microinjection of nor-BNI into the right CeA abolished stress-induced cutaneous allodynia in sumatriptan-primed rats; there were no significant reductions in periorbital or left hindpaw allodynia (Figure 4 (a) and (b)). Furthermore, microinjection of nor-BNI into the right CeA also blocked BLS-induced allodynia in the right hindpaw (Supplemental Figure S4 (a) and (b)). In contrast, microinjection of nor-BNI into the left CeA was not effective, as there were significant stress-induced reductions in periorbital (Figure 4 (a)) and either left or right hindpaw withdrawal thresholds (Supplemental Figure S4 (c) and (d)). Saline-primed rats showed no changes in withdrawal thresholds after BLS and microinjection of saline or nor-BNI. The integrated AOC of periorbital and hindpaw thresholds confirmed these results (Figure 4 (c) and (d)).
Lateralized effects of left or right CeA nor-BNI on BLS-induced cutaneous allodynia. (a), (b): BLS-induced periorbital (a) and hindpaw (b) allodynia was blocked by the administration of nor-BNI (2.5 µg/0.5 µl) into the right, but not left, CeA prior to the first BLS episode in sumatriptan pre-exposed rats. *p < 0.05, **p < 0.01, ***p < 0.001 vs. baseline (BL). Group sizes are shown on the figure. (c), (d): Analysis of Area Over the Curve (AOC) confirmed that BLS induced periorbital (c) and hindpaw (d) allodynia was blocked by administration of nor-BNI into the right, but not left CeA. *p < 0.05, **p < 0.01. Data are reported as the mean ± SEM. Statistics performed using two-way ANOVA followed by Dunnet’s multiple comparisons post-hoc test for (a) and (b); one-way ANOVA followed by Fisher’s LSD post-hoc test for (c) and (d).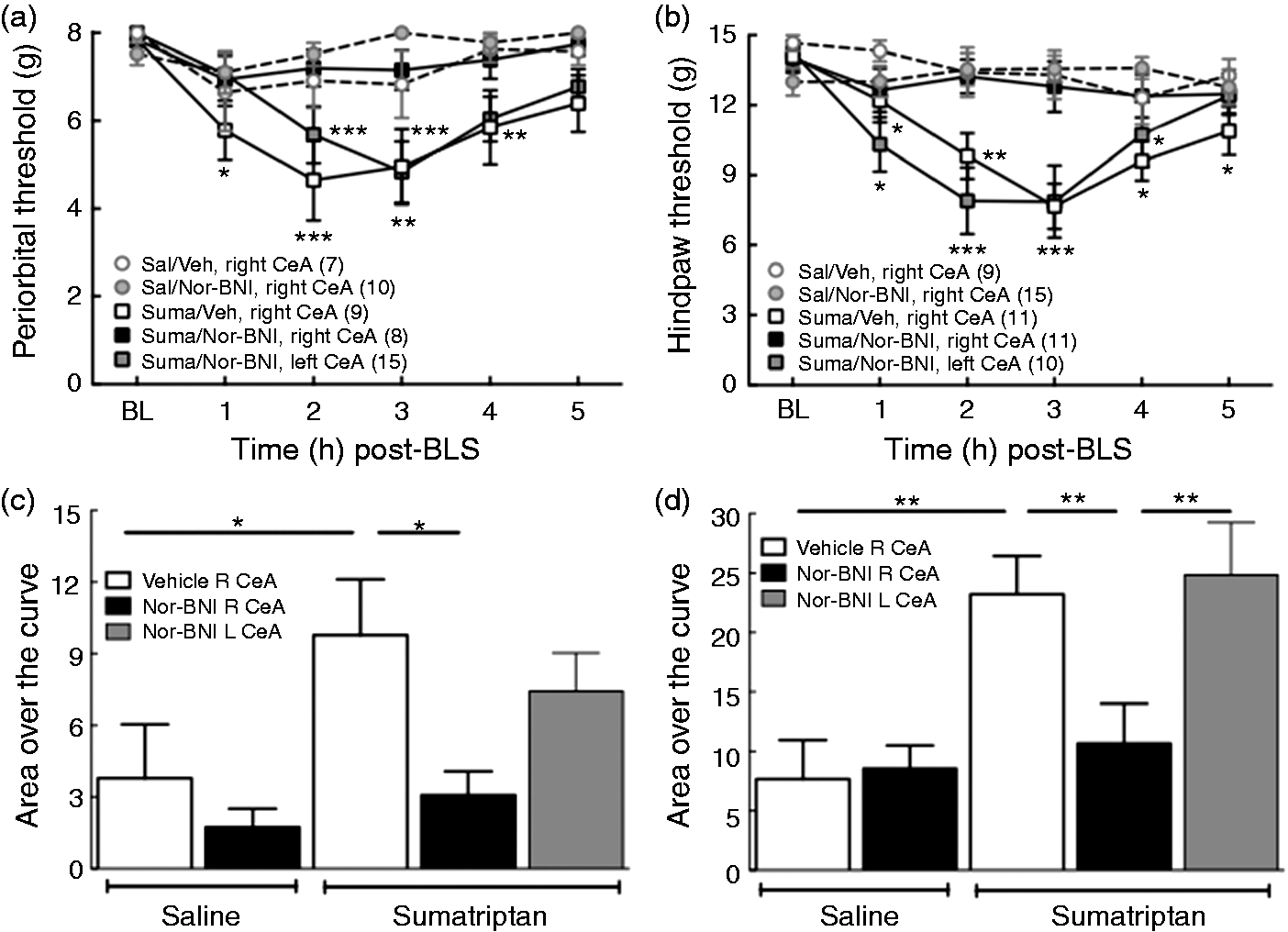
Right CeA administration of an MEK inhibitor, U0126, prevents BLS-induced cutaneous allodynia
We assessed whether U0126, an inhibitor blocking MEK signaling potentially downstream of KOR, could block BLS induced cutaneous allodynia in sumatriptan-primed rats. The microinjection of U0126 (1 µg) into the right CeA of sumatriptan-primed rats 15 min prior to the second BLS exposure significantly decreased both periorbital and hindpaw allodynia (Figure 5 (a) and (b)). U0126 microinjection blocked stress-induced elevation of AOC for periorbital (t(10) = 2.639; p = 0.0248) thresholds and approached, but did not meet, the criterion for statistical significance for hindpaw (t(23) = 2.052; p = 0.0517) withdrawal thresholds when compared to saline-microinjected rats (Figure 5 (c) and (d)).
CeA U0126, a MEK inhibitor, blocked BLS-induced cutaneous allodynia. (a), (b): Administration of U0126 (1 µg/0.05 µl) into the right CeA 15 min prior to the second BLS blocked periorbital (a) and hindpaw (b) allodynia in sumatriptan-primed rats. *p < 0.05 vs. baseline (BL). Group sizes are shown on the figure. (c), (d): Group comparisons of Area Over the Curve (AOC) for the periorbital (c) and hindpaw (d) allodynia demonstrated that the U0126 pretreatment group showed significantly less periorbital allodynia induced by BLS compared to vehicle treated rats (*p < 0.05) and reduced hindpaw allodynia that did not reach the significance cutoff (p = 0.0517). Data are reported as the mean ± SEM. Statistics performed using two-way ANOVA followed by Dunnett’s multiple comparisons post-hoc test for (a) and (b); unpaired t-test (two-tailed) for (c) and (d).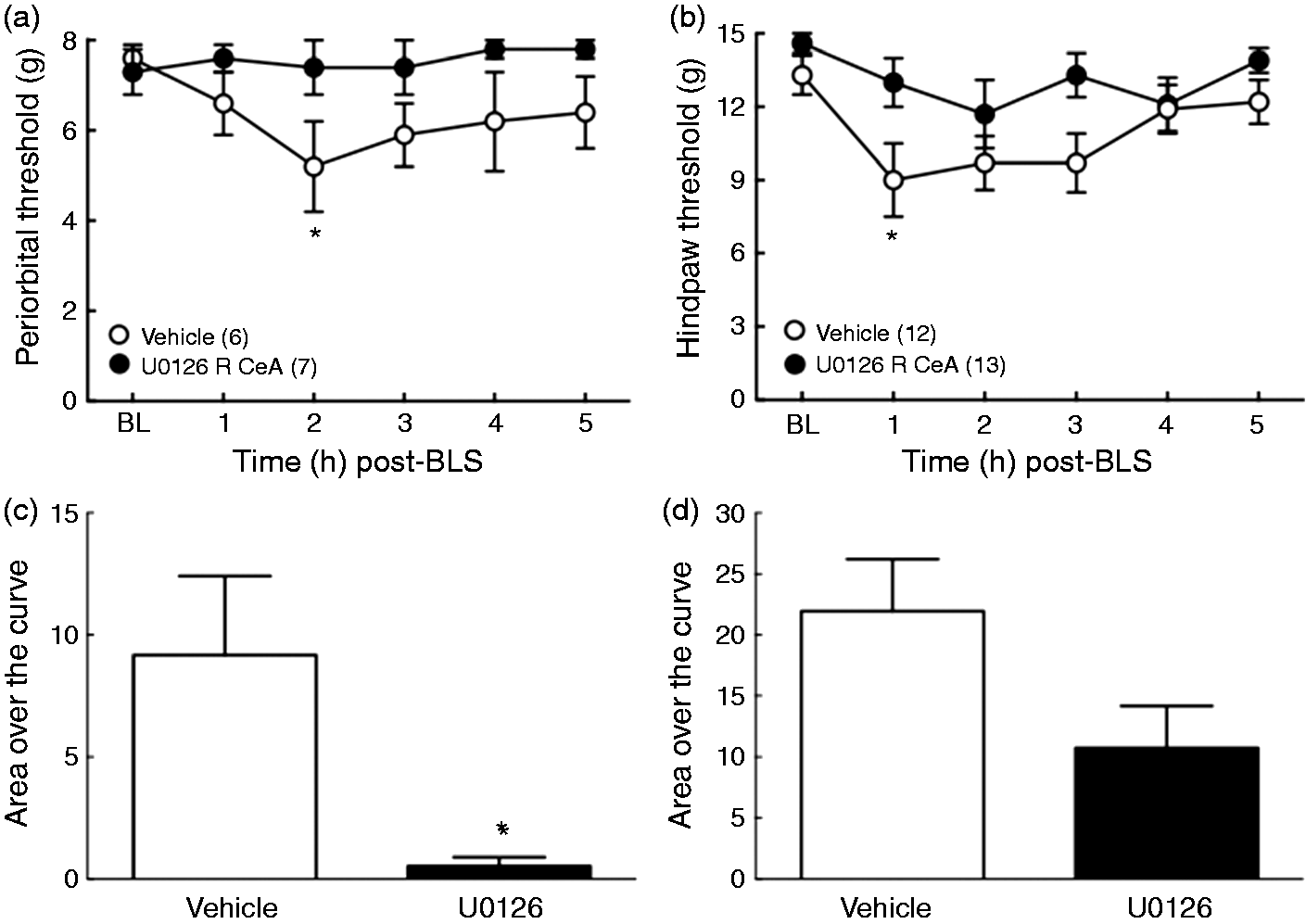
Right, but not left, CeA injection of the KOR agonist produced allodynia in sumatriptan-primed rats
We determined whether exogenous injection of a KOR agonist, U69,593, into the right CeA could induce cutaneous allodynia in saline- or sumatriptan-primed rats in the absence of stress (Figure 6). Microinjection of U69,593 (1 µg) into the right, but not left, CeA of sumatriptan, but not saline-primed rats produced a significant reduction in periorbital or hindpaw withdrawal thresholds at 40–60 min after administration (Figure 6 (a) and (b)). Microinjection of saline into the right CeA did not produce any changes in tactile thresholds. These results are reflected in the integrated AOC analysis (Figure 6 (c) and (d)). The AOC of sumatriptan-primed rats receiving right CeA U69,593 was significantly greater than those determined after U69,593 given to saline-primed rats for periorbital and hindpaw withdrawal thresholds. U69,593 did not produce periorbital or hindpaw allodynia following microinjection into the left CeA of sumatriptan-primed rats.
Cutaneous allodynia induced by U69,593, a KOR agonist, into the right, but not left, CeA of sumatriptan primed rats. (a), (b): Administration of 1 µg U69,593 into the right, but not left CeA produced periorbital (a) and hindpaw (b) allodynia in sumatriptan, but not saline-primed rats. *p < 0.05, ***p < 0.01 vs. baseline (BL). Group sizes are shown on the figure. (c), (d): Group comparisons of Area Over the Curve (AOC) for periorbital (c) and hindpaw (d) allodynia demonstrated that microinjection of U69,593 into the right, but not left CeA, produced tactile allodynia selectively in sumatriptan-primed rats. *p < 0.05, **p < 0.01. (e), (f): Right CeA administration of the MEK inhibitor, U0126 (1 µg), 5 min prior to U69,593 (1 µg, right CeA) blocked U69,593-induced periorbital (e) and hindpaw (f) tactile allodynia in sumatriptan-primed rats. *p < 0.05, **p < 0.01, **p < 0.001 vs. BL. Group sizes are shown on the figure. Data represent mean ± SEM. Statistics performed using two-way ANOVA followed by Dunnett’s multiple comparisons post-hoc test for (a), (b), (e) and (f); One-way ANOVA followed by Fisher’s LSD post-hoc test for (c) and (d).
Right CeA administration of U0126 blocked U69,593-induced allodynia in sumatriptan-primed rats
Administration of the MEK inhibitor, U0126 (1 µg), 5 min prior to U69,593 (1 µg) into the right CeA, abolished periorbital and hindpaw allodynia induced by right CeA U69,593 administration (Figure 6 (e) and (f)). These results suggest that U69,593-induced hindpaw allodynia in sumatriptan-primed rats is dependent on the MEK signaling cascade.
Discussion
In this study, we demonstrated that KOR antagonism prevented the expression of stress-induced cephalic and extracephalic allodynia in sumatriptan-primed rats. Moreover, KOR antagonists blocked BLS-induced elevation in plasma CGRP in sumatriptan-primed rats. The data revealed a novel, lateralized KOR-mediated circuit within the right CeA that may mediate stress-induced cutaneous allodynia and increase plasma CGRP. This dynorphin/KOR signaling pathway in the right CeA appears to play a pivotal role in mediating stress-related outcomes, opening a potential new path for the preventive treatment of migraine.
It is now appreciated that medications that are used for migraine therapy can themselves produce powerful adaptive changes in the brain. Overuse of medications that are prescribed for acute treatment of migraine, including triptans, can induce MOH, a condition characterized by increased frequency of migraine attacks. We have previously suggested that the increased sensitivity to putative migraine triggers in an animal model of MOH based on “reverse translation” could be due to decreased thresholds for activation of neural circuits, leading to the multiple phases of migraine (38,46,48). As noted above, treatment with sumatriptan for a one week period followed by a two week interval produced a long-lasting state of latent sensitization in which animals demonstrated hypersensitivity to putative migraine triggers, reflected by measures often observed clinically including cutaneous allodynia and increased plasma CGRP (38). The CGRP dependency of this model was demonstrated by prevention with an anti-CGRP antibody (36), again consistent with clinical observations that support a critical role of CGRP in migraine pathophysiology exemplified by increased levels of CGRP, but not substance P (SP) in the jugular blood (49); induction of migraine following the intravenous infusion of CGRP in migraineurs (50,51); and the clinical efficacy of blockade of CGRP signaling with small molecule CGRP receptor antagonists (52,53) or anti-CGRP antibodies (13,14). We emphasize, however, that like other preclinical models, our approach is not intended to directly replicate the human condition but provides a means to explore potential mechanisms that ultimately require clinical evaluation.
The injury-free model employed in these studies was based on clinical observation of MOH, and, for this reason, the relevance of conclusions to episodic or chronic migraine should be made cautiously. Nevertheless, the model allows for mechanistic investigation of brain circuits that could link putative migraine triggers to the migraine attack. Stress is the most commonly reported trigger of migraine, but the underlying mechanisms of how this occurs remain unclear. Stress-induced activation of the hypothalamic-pituitary-adrenal (HPA) axis has been shown in migraine patients. Imaging studies have demonstrated hypothalamic and limbic activation in migraineurs both during the migraine attack and in the interictal period (32). Resting state functional connectivity (rs-fc) between the hypothalamus and the autonomic circuits (54) or between the amygdala and the insula (55) were increased in migraine patients when compared to healthy controls. The central nucleus of the amygdala (CeA) is commonly referred to as the “nociceptive” amygdala and it is possible that outputs of the CeA to descending pain modulatory centers, including the periaqueductal gray (PAG) and rostral ventromedial medulla (RVM), could amplify stress-induced activation of meningeal nociceptors and promote pain. Thus, convergence of circuits in the hypothalamus and amygdala could link stress to both the premonitory and the pain phases of migraine. Notably, dynorphin-expressing neurons project from the hypothalamus to the amygdala, providing a possible stress-related functional link between these structures (56–58).
Chronic migraine is often comorbid with depression, and is associated with anxiety and negative cognitive function (for review, see (59)). The dynorphin/KOR system is now believed to be a therapeutic target for the treatment of stress-related mood disorders including anxiety, depression, drug seeking and relapse (20–23), effects are consistent with the distribution of dynorphin and the KOR throughout the limbic system (60–62). KOR activation has been documented to elicit dysphoria in humans (63) and has been shown to produce aversion in preclinical models (for reviews, see (62)), effects that may be due to inhibition of the mesocorticolimbic dopaminergic system (22,64,65). Stress is known to induce corticotrophin releasing factor (CRF) signaling (66–68). While blockade of KOR abolishes CRF-induced aversion and anxiety-like behaviors, blockade of CRF receptors does not abolish aversion or anxiety-related behaviors induced by KOR agonists, indicating that KOR activation occurs “downstream” from CRF activation (21,22). Such observations suggest that stress could activate a CRF to dynorphin to KOR pathway from the hypothalamus and other brain regions important to the stress responses (21).
Our data demonstrate that environmental stress produces delayed, generalized and sustained cutaneous allodynia selectively in animals with sumatriptan-induced latent sensitization. It should be noted that the allodynia peaks at approximately two hours after the BLS, possibly suggesting “relief” of stress, also reflecting the delay frequently seen between the onset of stress and emergence of migraine in humans. The delayed onset relevant to stress is also consistent with the time necessary for engagement of stress-relevant cascades in pain modulatory circuits. The period of exposure to the BLS and the delayed maximal allodynia may allow for sufficient time for engagement of stress related pathways, including the release of sympathetic stress mediators and activation of central dynorphinergic circuits.
In our studies, KOR antagonists given either during sumatriptan priming or immediately prior to BLS prevented the expression of BLS-induced cutaneous allodynia and increase in plasma CGRP. Three injections of nor-BNI during the sumatriptan priming period fully prevented the stress-induced allodynia and increases in plasma CGRP, suggesting that KOR activation might play a role in promoting sumatriptan-induced latent sensitization. However, nor-BNI has been shown to have an extremely long duration of action in preclinical models (69,70). This administration schedule might promote partial KOR blockade even two weeks after termination of drug administration. For this reason, we evaluated administration of the short-acting KOR antagonist, CYM51317, administered immediately before the two episodes of BLS and showed virtually complete prevention of BLS-induced allodynia in sumatriptan-primed rats. These observations suggest that antagonist occupation of KOR is likely important in the expression of stress-induced consequences, including cephalic and extracephalic allodynia and increased plasma CGRP. The possibility that occupation of the KOR plays a role in the development of latent sensitization following sumatriptan cannot be excluded and requires further investigation.
The role of stress-related central circuits was investigated further by determining whether KOR signaling from the amygdala could promote hyperalgesic responses in rats with latent sensitization. Elevated levels of dynorphin and phosphorylated KOR were detected in both the right and left amygdala of sumatriptan-primed rats. However, KOR blockade in the right, but not the left, CeA blocked stress-induced cutaneous allodynia regardless of whether allodynic thresholds were measured in the right or left hindpaws. Consistent with a hyperalgesic output from the right but not left CeA, microinjection of U69,593 into the right, but not left, CeA produced cephalic and extracephalic allodynia selectively in sumatriptan-primed rats. The involvement of KOR signaling was further supported by the demonstration that microinjection into the right CeA of U0126, an inhibitor of the potential KOR downstream kinase MEK, blocked both stress and U69,593-induced cephalic and extracephalic allodynia. These data are consistent with the blockade of BLS-induced cutaneous cephalic and extracephalic allodynia observed following CeA microinjection of nor-BNI in the right, but not left, amygdala.
There are lateralized responses in amygdala nuclei in diverse biological responses such as, for example, predator-induced fear responses (71). Multiple groups have reported that the right, but not left, amygdala is activated in pain states. A functional MRI (fMRI) study showed that visceral pain (gastric fundus distention) selectively activated the right amygdala in humans (72). Preclinical studies have shown potentiated efferent activity from the right amygdala to the PAG during predator-induced stress (i.e., rats exposed to cats) (73). Electrophysiological recordings showed activation of right amygdala neurons by either left or right knee arthritis (74) or following induction of experimental neuropathic pain by injury to the sciatic nerve in either the right or left hindlimb (75). Thus, our finding of the lateralization of the CeA-mediated stress response to the right amygdala is consistent with these previous reports, and now highlights the functional importance of KOR signaling in the CeA. Even though dynorphin and phosphorylated KOR levels were increased by stress in both the right and left amygdala, only KOR signaling from the right CeA appears to promote hyperalgesia in rats with latent sensitization. The underlying circuits that mediate lateralization remain to be explored.
Conclusion
Mounting clinical and preclinical evidence supports the role of dynorphin/KOR system in promoting stress-related disorders (20–23). Stress is a known clinical trigger of migraine attacks (17,76) and our data suggest that KOR antagonists could provide a new therapeutic strategy for migraine prevention. JD-Tic is a KOR antagonist that has progressed to humans, where cardiovascular toxicity halted further development (77). It is possible, however, that this side-effect was compound, rather than mechanism, based, and that novel orally active and safe KOR antagonists can be discovered and progress to clinical trials. If effective, such compounds would offer the possibility of novel preventive migraine therapy with convenient administration, safety and reversibility.
Article highlights
Kappa opioid receptor (KOR) antagonists given systemically prevented stress-induced cephalic and extracephalic allodynia as well as plasma CGRP release in a model of MOH cephalic pain induced by sumatriptan priming. Blockade of KOR signaling in the right, but not left, central nucleus of the amygdala inhibited stress-induced cephalic and extracephalic allodynia in sumatriptan-primed rats. KOR antagonists may represent a novel class of medications for migraine prevention.
Footnotes
Acknowledgement
We thank Dong Lu and Carlos Eduardo da Silva Monteiro for their technical support. ER, HR, DD and FP conceived and planned the study; JYX, MDF, CMK, NME, JL, BR, XY, PH and NG performed the experiments; JYX, MDF, TK, MO, XY, EN and JS analyzed the data; JYX, TK, MO and FP wrote the manuscript; DD, ER, HR, MDF, EN, CMK, BR, and JS corrected and improved the manuscript.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NS 069575 (F.P.).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.