
Abstract
Objective
To review the role of CGRP in human models of primary headaches and to discuss methodological aspects and future directions.
Discussion
Provocation experiments demonstrated a heterogeneous CGRP migraine response in migraine patients. Conflicting CGRP plasma results in the provocation experiments are likely due to assay variation; therefore, proper validation and standardization of an assay is needed. To what extent CGRP is involved in tension-type headache and cluster headache is unknown.
Conclusion
Human models of primary headaches have elucidated the role of CGRP in headache pathophysiology and sparked great interest in developing new treatment strategies using CGRP antagonists and antibodies. Future studies applying more refined human experimental models should identify biomarkers of CGRP-induced primary headache and reveal whether CGRP provocation experiments could be used to predict efficacy of CGRP antagonists in migraine patients.
Introduction
Human headache models provide a unique opportunity to investigate the underlying pathophysiology of primary headaches. One of the most extensively studied signaling molecules is calcitonin gene-related peptide (CGRP), which is widely distributed in the nervous system (1–4), particularly in the perivascular trigeminal sensory afferents (5) and the trigeminal nucleus caudalis (6). CGRP dilates human cerebral arteries (7,8), induces mast cell degranulation (9) and plasma extravasation (10), and is released upon trigeminal ganglion thermocoagulation (11). Randomized clinical trials (RCT) have demonstrated the efficacy of CGRP antagonists and monoclonal antibodies in migraine treatment (12–17). In addition, phase III RCTs in migraine are ongoing and RCTs on CGRP antibodies are initiated in cluster headache patients (ClinicalTrials.gov: NCT02438826 and NCT02397473).
CGRP has been studied directly as a trigger of migraine attacks and indirectly by measuring CGRP plasma levels after triggering primary headaches using glyceryl trinitrate (GTN) and pituitary adenylate cyclase activating peptide 38 (PACAP38) infusion. Here, we review these studies, discuss methodological aspects, and outline future directions.
CGRP in the GTN headache model
GTN is a nitric oxide (NO) donor, which serves as a tool to investigate the role of NO in primary headaches. The GTN headache model was developed and validated through extensive human experimental studies (18). Juhasz et al. (19) investigated CGRP plasma levels during GTN-induced migraine attacks and reported increased CGRP levels during induced migraine attacks in 10 patients, but found no CGRP changes in five patients without induced attacks. In those who developed attacks, blood samples were collected one to two hours after the beginning of the migraine-like attacks, which started five to seven hours after GTN infusion. In patients who did not develop attacks, blood samples were taken five to six hours after GTN infusion. Sumatriptan nasal spray, administered 120 minutes after the onset of the induced attack, decreased CGRP levels and ameliorated headache (20). The authors suggested that GTN infusion activates trigeminal nerve fibers to liberate CGRP and thereby induce migraine attacks.
In an RCT study, migraine patients received a GTN infusion on two separate days, followed by either intravenous infusion of the CGRP receptor antagonist olcegepant 10 mg or a placebo (21). The majority of migraine patients developed the usual biphasic headache pattern after GTN infusion: Mild to moderate headache during an immediate phase within the first 60 minutes after infusion, and migraine-like attacks during a delayed phase in the next 2–12 h after infusion. Olcegepant or placebo was administrated one hour after the start of infusion. Olcegepant failed to prevent GTN-induced migraine (seven patients reported migraine after olcegepant compared to nine patients after placebo). Interestingly, CGRP plasma levels were unchanged at baseline, 30, 60, 90, 120, 180 and 240 minutes (21), which is in contrast to the study by Juhasz et al. (19) reporting elevated CGRP plasma levels at one to two hours after onset of delayed migraine-like attacks approximately five hours after GTN infusion. The lack of efficacy of olcegepant in prevention of GTN-induced migraine could have three possible explanations: 1) CGRP acting upstream from NO in the migraine-generating cascade (21); 2) CGRP pathways are independent from NO migraine-inducing pathways; 3) Olcegepant terminates acute attacks of migraine, but is not effective in preventing attacks.
In tension-type headache, CGRP changes were examined in blood samples collected from the antecubital vein during GTN-induced headache in 16 chronic-tension-type headache (CTTH) patients and 16 controls (22). The study showed that GTN-induced immediate headache was not associated with elevated plasma CGRP in CTTH.
In cluster headache (CH), Fanciullacci et al. investigated CGRP levels in the external jugular vein during sublingual nitroglycerine-induced cluster attacks (23). Baseline CGRP plasma levels were higher in the patients who were in an active period compared to a remission period (23). Only cluster headache patients during an active period reported nitroglycerine-induced attacks (23,24) associated with further increase in plasma CGRP that reversed following sumatriptan injection (24). Based on these findings, the authors proposed CGRP as a marker for activation of the trigeminovascular system in cluster headache (23,24).
CGRP migraine models
Lassen et al. (25) reported the first evidence on CGRP provoking migraine attacks. Twelve migraine without aura (MO) patients were randomly allocated to CGRP or placebo infusion over 20 minutes. Originally, the authors strictly applied the International Headache Society classification criteria for MO (26) and reported migraine in three out of nine patients. Later, different criteria have been applied in experimentally-induced migraine attacks (27), since migraine attacks experimentally provoked by pharmacological substances are not spontaneous and, therefore cannot fulfill the strict IHCD criteria for migraine without aura (28). By applying these criteria, we calculated that 67 % of the MO patients experienced delayed migraine-like attacks compared to one in the control group. Interestingly, CGRP-induced migraine-like attacks had a similar time of onset as reported in a GTN headache provocation study (29).
CGRP provocation experiments have also been extensively studied in migraine with aura (MA). Hansen et al. (30) investigated migraine patients suffering exclusively from MA attacks. After CGRP infusion, 57 % of patients reported migraine-like attacks without aura (30). In addition, four out of 14 reported typical aura symptoms, which were likely due to experimental stress. It would be interesting to re-challenge these patients with CGRP. Familial hemiplegic migraine (FHM) is an autosomal dominant subtype of MA (31–32), and provocation studies showed that CGRP did not provoke migraine attacks in FHM patients with (33) and without (34) known mutations. These data suggest a different neurobiological pathway in MO and MA compared with FHM, which renders FHM patients less CGRP sensitive than in the common migraine subtypes. Interestingly, pre-clinical studies showed a decreased population of CGRP-immunostained cells (35) and increased CGRP release (36) in the trigeminal ganglion in FHM1 knock-in mice compared with normal wild-type mice. Taken together, provocation studies demonstrated that the majority of MO and MA patients developed delayed migraine-like attacks after CGRP infusion. This is indicative of a CGRP involved common cascade initiating migraine pain in both MO and MA patients.
The CGRP vascular effects in relation to migraine have been studied by ultrasound-based imaging and brain imaging. Two studies reported that CGRP dilated the superficial temporal artery measured by two-dimensional ultrasound and the middle cerebral artery (MCA) measured indirectly via Doppler measured MCA velocity (13,37). Asghar et al. examined intra- and extracerebral vessels after CGRP infusion in healthy volunteers using high-resolution magnetic resonance angiography (MRA) (38). CGRP dilated the extracranial part of the middle meningeal artery (MMA), but in contrast to Doppler MCA velocity data, did not dilate the MCA. This study also showed that sumatriptan constricted the MMA but not the MCA (38). A functional magnetic resonance imaging (fMRI) study investigated the blood oxygenation level-dependent (BOLD) response, as a surrogate marker of neuronal activity in the visual cortex after CGRP and placebo infusion (39). This study found that CGRP infusion did not modulate neuronal activity in the brain – likely due to exogenous CGRP not crossing the BBB. However, following CGRP infusion in healthy volunteers, noxious heat stimuli applied in the trigeminal ophthalmic region resulted in an increased BOLD response in the brainstem and insula compared with baseline stimulation (40). In addition, decreased BOLD response was found in the thalamus, cingulate cortex, and caudate nuclei (40). Subcutaneous injection of sumatriptan reversed these changes in BOLD response (40). These data indicate that systemic CGRP might modulate nociceptive transmission in the trigeminal pain pathway. Finally, an MRA study in MO patients examined circumference changes in the extracranial part of the MMA and MCA during CGRP-induced delayed migraine attacks (41). CGRP induced delayed migraine-like attacks were associated in 75% of the patients with dilation of the MMA and MCA on the headache side. Subcutaneous injection of sumatriptan caused cessation of the migraine-like attacks and constricted only the MMA, whereas sumatriptan had no effect on the MCA. These findings suggested that constriction of the MMA and not the MCA were associated with amelioration of headache. A similar effect of sumatriptan on the MMA circumference was reported during spontaneous migraine attacks (42). Collectively, these data suggest that CGRP is unlikely to cross the blood brain barrier (BBB), suggesting a peripheral site of action. In support, a recent study reported that CGRP induced migraine-like attacks were not associated with premonitory symptoms (a marker of CNS involvement) (43). Provocation studies also revealed a heterogeneous response, i.e. some patients developed attacks while others did not. The question is to what extent a high family load of migraine susceptibility genes would affect hypersensitivity to CGRP. A recent study addressed this hypothesis and reported no association between high family load of migraine susceptibility genes and hypersensitivity to CGRP infusion (44). Thus, the conferring risk of MO by high family load had no additive effect on CGRP-induced migraine-like attacks.
CGRP in the PACAP38 model of migraine
PACAP38 is widely distributed in the human nervous system (45). In rats, locally applied PACAP38 caused CGRP release from the trigeminal nucleus caudalis, but not from the trigeminal ganglion (46). PACAP38 provokes migraine attacks in 58–73 % of MO patients (47,48) and dilates extracerebral arteries, but not intracerebral arteries (47) assessed by MRA. A recent study investigated CGRP levels after PACAP38 provocation (49). Patients were stratified into two groups: Those who developed migraine-like attacks and those who did not. CGRP plasma levels were measured several times, lastly 90 minutes after the start of infusion. Between the two patient groups, no differences in CGRP levels were found. It seems that PACAP38 administration did not cause immediate CGRP release in the blood. It should be noted that possible local CGRP release from the trigeminal nucleus caudalis and trigeminal ganglion are unlikely to be detected by collected blood samples. Furthermore, whether CGRP is increased during PACAP38-induced attacks remains unknown.
Discussion
The present review has identified several methodological aspects important to discuss for future studies. In the following, we will discuss: 1) Plasma CGRP as a biochemical marker in human experimental models; 2) heterogeneity of the CGRP response in migraine patients; 3) brain imaging biomarkers of CGRP-induced primary headache attacks; 4) future experimental models using CGRP.
CGRP as a biochemical marker
Overview of reported CGRP plasma level changes in experimental headache provocation studies.
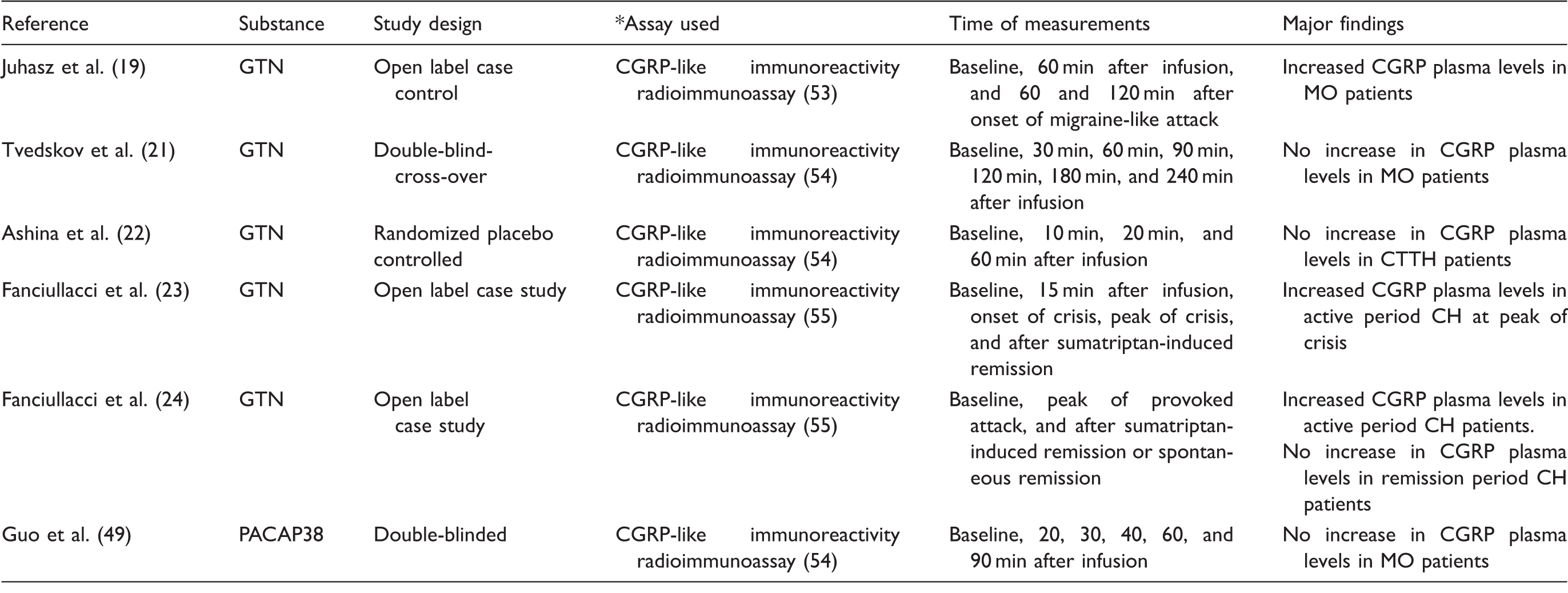
Migraine without aura (MO); Cluster headache (CH); Chronic tension-type headache (CTTH); Glyceryl trinitrate (GTN); Pituitary adenylate cyclase activating peptide 38 (PACAP38).
Some GTN provocation studies reported a triptan-induced decrease in CGRP plasma levels. The exact underlying mechanism remains unclear. The triptan-induced decrease in CGRP release might be due to a direct inhibition via presynaptic 5-HT receptors on trigeminal nerve endings (50). Alternatively, it could be secondary to a decreased activation of perivascular trigeminal nerves because of cranial vasoconstriction. Separating neuronal and vascular mechanisms of action has proven very difficult so far, and is a limitation in human in vivo research. Therefore, it remains to be proven if and how triptans affect CGRP plasma concentrations during an acute migraine attack.
One of the major methodological reservations in the triptan and CGRP interaction studies is that none of the GTN provocation studies were placebo-controlled (20,23,24). Thus, CGRP levels may decrease spontaneously during a migraine attack. It would be interesting to investigate CGRP levels during the full course of spontaneous migraine attacks. During untreated GTN-induced migraine attacks, CGRP levels decrease after an initial increase within the first hour after the onset of attacks (19). Also, the GTN provocation studies collected blood samples at different time points after the start of infusion (19,21,22,23,24). The study by Juhasz et al. (19) collected blood samples several hours after the onset of attack. Therefore, CGRP levels might already have been decreasing spontaneously at the time of triptan administration in other studies. If we assume that sumatriptan decreases CGRP levels, it is likely because they were elevated prior to administration. In support, sumatriptan did not cause changes in plasma CGRP at baseline in healthy volunteers (51).
Heterogeneity of the CGRP response, brain imaging, and future studies
The headache-inducing mechanisms of CGRP are not fully clarified. Migraine studies have demonstrated that 68 % of MO and MA patients are responders to CGRP infusion, i.e. these patients reported migraine-like attacks (25,30,41). It is unknown why 32% of patients did not develop migraine attacks, and the question is whether these patients would respond to CGRP antagonists. It should be noted that fluctuating susceptibility to migraine (52) might influence the CGRP response. The number of MA patients who reported delayed headache was significantly higher than delayed headache reported by controls (30). It is possible that some patients with delayed headache did not develop full-blown migraine attacks because of fluctuating susceptibility to migraine. To date, no study has been conducted to reproduce the lack of CGRP response in the same population of non-responders. It would be interesting to investigate the efficacy of CGRP antagonists and antibodies in a migraine population that has been phenotyped as CGRP infusion responders versus non-responders. This would enable us to possibly predict the efficacy of future drugs targeting CGRP in patients.
Brain imaging studies have shown that CGRP causes MMA dilation (38) and modulates nociceptive transmission in deep brain structures (40). It is not fully clarified whether CGRP induced MMA dilatation is an epiphenomenon or important for the development of migraine headache. Future studies should examine whether CGRP antibody treatment blocks CGRP-induced MMA dilation and migraine-like attacks.
Considering that CGRP levels are increased after GTN in cluster headache provocation experiments (24), there is a need to examine whether CGRP induces cluster headache attacks. Such an experiment would also be interesting to conduct on CTTH patients, since no CGRP plasma increase has been reported in these patients (22). PACAP38 infusion did not change the plasma levels of CGRP in migraine patients (49), but it is unknown whether PACAP38-induced migraine attacks can be aborted or prevented by CGRP antagonism. An ongoing clinical trial is investigating the preventative effect of AMG334 in the PACAP38 migraine model (ClinicalTrials.gov: NCT02542605).
Conclusion
Human models of CGRP have contributed significantly to elucidating CGRP’s role in the pathophysiology of primary headaches, particularly in migraine. It remains unclear to what extent CGRP is involved in TTH and cluster headache. Future studies using more refined human experimental models including brain imaging should investigate the site of action of CGRP-induced headache to identify specific biomarkers.
Article highlights
Provocation experiments demonstrated a heterogeneous CGRP migraine response in migraine patients. Conflicting CGRP plasma results in the provocation experiments are likely due to assay variation, and therefore proper validation and standardization of an assay is needed. To what extent CGRP is involved in tension-type headache and cluster headache is unknown. Future experiments might reveal whether CGRP provocation experiments could be used to predict the efficacy of CGRP antagonists in migraine patients.
Footnotes
Declaration of conflicting interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: M Ashina is a consultant or scientific advisor for Allergan, Amgen, Alder, ATI and Eli Lilly, and a primary investigator for Amgen 20120178 (Phase 2), 20120295 (Phase 2), 20130255 (OLE), 20120297 (Phase 3) and GM-11 gamma-Core-R trials. H Ashina and H Schytz report no disclosures relevant to the manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by The Lundbeck Foundation (R155-2014-171), the Novo Nordisk Foundation (NNF11OC1014333), the University of Copenhagen, the Research Foundation of the Capital Region of Denmark (A4620), the Danish Council for Independent Research-Medical Sciences (DFF-4004-00169B), Independent Research-Medical Sciences (FSS) (DFF-1331-00210A) and the FP7-EUROHEADPAIN (602633).