
Abstract
Background
This study explored whether antagonism of orexin receptors might be an effective mechanism for migraine prevention.
Methods
We conducted a randomized, double-blind, placebo-controlled, pilot trial. Patients experiencing four to 14 days with migraine during a one-month baseline period were randomized to the orexin receptor antagonist filorexant 10 mg nightly or placebo for three months. Efficacy was assessed by mean monthly migraine days (headache plus at least one associated migraine symptom) and headache days. Safety and tolerability were assessed by adverse event reports and laboratory tests.
Results
Of 120 patients treated with filorexant and 115 treated with placebo, 97 (81%) and 101 (88%), respectively, completed the trial. There was no statistically significant difference between treatments for change from baseline in mean monthly migraine days (filorexant = −1.7, placebo = −1.3, difference = −0.4 (95% CI: −1.3, 0.4)) or headache days (filorexant = −1.7, placebo = −1.2, difference = −0.5 (95% CI: −1.4, 0.4)). Filorexant was generally well tolerated but was associated with a higher proportion of patients who reported adverse events than placebo (47% vs 37%), particularly somnolence (13% vs 4%).
Conclusions
These data fail to provide evidence that antagonism of orexin receptors with filorexant, when administered at night, is effective for migraine prophylaxis.
Introduction
There has been major progress in the acute pharmacological treatment of migraine in recent years with the development of the triptans (1), and promising current research focused on antagonism of calcitonin gene-related peptide (CGRP) receptors (2) or 5HT1F receptor agonists (3). However, there has been relatively little progress in developing migraine-specific preventive treatments for frequent episodic migraine. Estimates indicate that approximately 25% of migraine patients are candidates for preventive therapy but a substantial proportion of these do not receive prophylaxis (4).
The orexin (hypocretin) signaling system, which originates in the hypothalamus, was discovered in the late 1990s (5,6) and has been evaluated primarily with regard to its role in regulating wakefulness (7). Orexin receptor antagonists such as almorexant and suvorexant have been shown to be effective for the treatment of insomnia (8,9). A role for orexin antagonists in headache disorders has also been postulated (10). Migraine and sleep are intimately interconnected (11). Sleep is both a trigger (e.g. sleep deprivation or excessive sleep) (12) and a remedy for migraine (13,14). Orexin neuropeptides are also thought to play a role in nociception. In rats, orexin receptor activation can modulate responses of the trigeminovascular system to dural stimulation (15–17). Furthermore, an orexin receptor antagonist showed efficacy in a rat model of trigeminal activation that has been predictive of clinical efficacy for migraine (17). Taken together, these observations suggest the possibility that antagonism of orexin receptors might offer a novel approach to migraine prevention.
The aim of the present exploratory phase 2 study was to evaluate the efficacy and safety of the orexin receptor antagonist filorexant (MK-6096) for migraine prophylaxis (18–20). A 10-mg dose was selected for evaluation because this dose demonstrated significant improvements in sleep onset and maintenance in a phase 2 dose-ranging study in patients with insomnia (21,22). Because of its demonstrated sedative effects, filorexant was given at night (just before bedtime).
Methods
Full details of the methods are provided in the study protocol (see supplementary material).
Patients
Patients were eligible for the study if they were 18–65 years of age, had migraine with or without aura for more than one year (23), experienced an average of four to 14 migraine days per month in the three months prior to screening, and four to 14 migraine days during the four-week baseline period prior to randomization.
Exclusion criteria
Patients were excluded if they had ≥15 headache days per month or had taken medication for acute migraine or other headaches on >10 days per month in any of the three months prior to screening. Patients taking migraine-preventive medications in the 30 days prior to screening were excluded. Patients with a history of narcolepsy, cataplexy, circadian rhythm disorder, parasomnia, sleep-related breathing disorder, restless legs syndrome, periodic limb movement disorder, excessive daytime sleepiness or difficulty sleeping due to a medical condition were excluded. Patients who had traveled across >3 time zones or engaged in shift work within the past two weeks, or who anticipated needing to travel across >3 time zones at any time during the study, were excluded. Patients were also excluded if they had a myocardial infarction, unstable angina, coronary artery bypass surgery, or other revascularization procedure, stroke, or transient ischemic attack within three months of screening. Patients with aspartate aminotransferase (AST), alanine aminotransferase (ALT), or bilirubin levels that were >1.5 × the upper limit of normal or serum creatinine that was >2 × the upper limit of normal at a laboratory screening visit were also excluded.
Regulatory and ethical matters
The study was conducted in accordance with principles of Good Clinical Practice and was approved by the appropriate institutional review boards and regulatory agency. Each patient provided written informed consent. The study was registered at ClinicalTrials.gov (NCT01513291).
Study design
This randomized, double-blind, placebo-controlled, parallel-group study (Merck Protocol 020) was performed at 19 investigative sites in the United States from February 2012 to October 2012. Study eligibility was assessed at a screening visit, following which eligible patients entered a four-week single-blind placebo observation period to establish baseline migraine frequency. Patients experiencing four to 14 migraine days during the four-week baseline period were randomized in a 1:1 ratio to filorexant 10 mg (two 5-mg tablets) or placebo (two matching tablets) nightly through a three-month treatment period. Patients who completed the entire three months of treatment entered a two-week double-blind run-out to evaluate the effects of abruptly stopping treatment. During the run-out, patients who received filorexant in the prior treatment period were randomized to filorexant or placebo in a 1:1 ratio while those who previously received placebo continued to receive placebo.
Patients were allocated in a double-blind fashion using a computer-generated randomized allocation schedule prepared by a blinded statistician at Merck. Randomization was stratified by monthly migraine days (standardized to 28 days) during the baseline period (≤ or >8 migraine days) and whether the patient was defined as having insomnia (based on a Sleep and Insomnia Assessment at screening). Numbered containers were used to implement allocation. Personnel at each study site used a central interactive voice response system to determine which container should be given to which patient. All study personnel, including the investigators, study site personnel, patients, and Merck staff remained blinded to treatment allocation throughout the study. Unblinding took place after data collection was complete.
Procedure
Patients were required to complete an electronic headache and sleep diary each day of the baseline period, during the treatment period, and for two weeks after the last dose of study medication, regardless of whether they experienced a headache. Patients were required to report headache information each night before bedtime and sleep information each morning on awakening. The following headache information was recorded daily by the patient: date, administration of study medication, use of any acute headache medication, presence of aura or other associated migraine symptoms, if headache duration was ≥30 minutes, maximum pain intensity, and any adverse experiences. The following sleep information was recorded: subjective total sleep time (sTST), subjective time to sleep onset (sTSO), subjective number of awakenings (sNAW), and subjective wake after sleep onset (sWASO). Patients returned to the clinic for planned follow-up visits at four, eight, 12, and 14 weeks post-randomization.
Safety was assessed by adverse event reports and routine laboratory tests performed at clinic visits including hematology, chemistry and urinalysis, electrocardiography (ECG), and vital sign assessments. A program-wide guidance document containing definitions of pre-specified events of clinical interest (ECI) was provided to investigators. Any adverse events judged by the investigator to be potentially suggestive of either cataplexy (e.g. falls) or sleep-onset paralysis were to be sent for adjudication but in practice no events of this type occurred.
Measures
A headache was defined as headache pain of ≥30 minute duration or headache of any duration for which acute treatment was administered. A headache day was defined as any occurrence of headache that started, ended, or recurred within a calendar day. Pain persisting for more than one calendar day after initial onset was considered as a new, distinct headache day. A migraine was defined as a headache with at least one associated symptom, i.e. aura, photophobia, phonophobia, nausea, or vomiting. A migraine day was defined as any occurrence of migraine headache that started, ended, or recurred within a calendar day. Pain persisting for more than one calendar day after initial onset was considered as a new, distinct migraine day. A migraine attack was defined as any migraine headache(s) that occurred within two consecutive calendar days. Pain persisting for more than two days after initial onset was considered a new distinct attack.
Statistical analyses
The primary efficacy endpoint was the change from baseline in mean monthly migraine days averaged over the three-month treatment period. The primary efficacy analyses used a full analysis set approach. All patients who were randomized, took at least one dose of study medication, and had at least one efficacy measurement were included in the treatment group to which they were randomized. The primary efficacy hypothesis was tested using a constrained longitudinal data analysis method (24) that included both baseline and post-baseline mean monthly migraine days for each month as response variables. The treatment group, time, the interaction of time by treatment, and monthly migraine days during the baseline period (≤ or >8 migraine days) were included in the model as categorical covariates, with a restriction that the baseline mean responses were the same across treatment groups (due to randomization). The treatment difference in terms of mean change from baseline in monthly migraine days to the post-baseline treatment period was estimated and tested from this model. The model allowed the inclusion of patients who were missing either the baseline or post-baseline measurements.
Secondary efficacy endpoints were: 1) the change from baseline in the mean monthly headache days averaged over the three-month treatment period, 2) the proportion of patients with at least a 50% reduction in mean monthly migraine days averaged over the three-month treatment period as compared to baseline, and 3) the proportion of patients with at least a 30% reduction in mean monthly migraine days averaged over the three-month treatment period as compared to baseline. Exploratory efficacy endpoints included: 1) the change from baseline in mean monthly migraine attacks averaged over the three-month treatment period, 2) the change from baseline in number of days per month requiring rescue medication averaged over the three-month treatment period, and 3) the change from baseline in sleep endpoints at Month 1, Month 2, and Month 3. Treatment response as defined by ≥50% and ≥30% reduction in mean monthly migraine days were analyzed based on a generalized linear mixed model using GLIMMIX. The GLIMMIX model included the fixed effects of treatment, time, treatment-by-time interaction, and monthly migraine days during the baseline period (≤ or >8 migraine days). Analysis of the other secondary and exploratory endpoints used the same methods as for the primary analyses. To control for multiplicity the formal testing of the one primary hypothesis and three secondary hypotheses was conducted in a sequential fashion, with each hypothesis test to be formally conducted only if the preceding test was rejected at the α = 0.05 level.
For the purpose of calculating the mean monthly rate data for the above analyses, one month was defined as 28 days. The mean monthly migraine days for each patient was calculated by dividing the total number of recorded headache days during the period by the number of days with diary records during that period and multiplying by 28. A minimum of seven days of diary data during the time period was required in order for the mean monthly rate to be calculated.
Tolerability and safety were summarized and assessed by a statistical and clinical review of all safety parameters, including adverse events, laboratory values, ECGs, and vital signs. The primary approach for all safety analyses was the all-patients-as-treated approach. All patients who received at least one dose of study therapy were included in the treatment group according to the treatment they received. The primary safety analysis was conducted on adverse events occurring within 14 days of dosing.
The data were analyzed using the SAS version 9.3 software package (SAS Corporation, Raleigh, NC, USA).
Power
The sample size was calculated based on a previous preventive study with topiramate (25) as well as data from a prophylaxis study with a CGRP receptor antagonist (26). With a planned sample size of approximately 220 randomized patients (110 in each group), the study had about 80% power to demonstrate the primary hypothesis that filorexant is superior to placebo in reducing the mean monthly migraine days averaged over three months of treatment, assuming a treatment difference of 1.6 migraine days with an SD of 4.1.
Results
Patient accounting and demographics
The trial flowchart is shown in Figure 1. Of the 120 patients treated with filorexant and 115 treated with placebo, 97 (81%) and 101 (88%), respectively, completed the three-month treatment period. There was no single predominant reason for the increase in discontinuations in the filorexant group compared to placebo. All patients who completed the three-month treatment period entered the two-week run-out and all but two completed the run-out.
Study flowchart.
Baseline characteristics of treated patients: number (%) of patients except where stated.
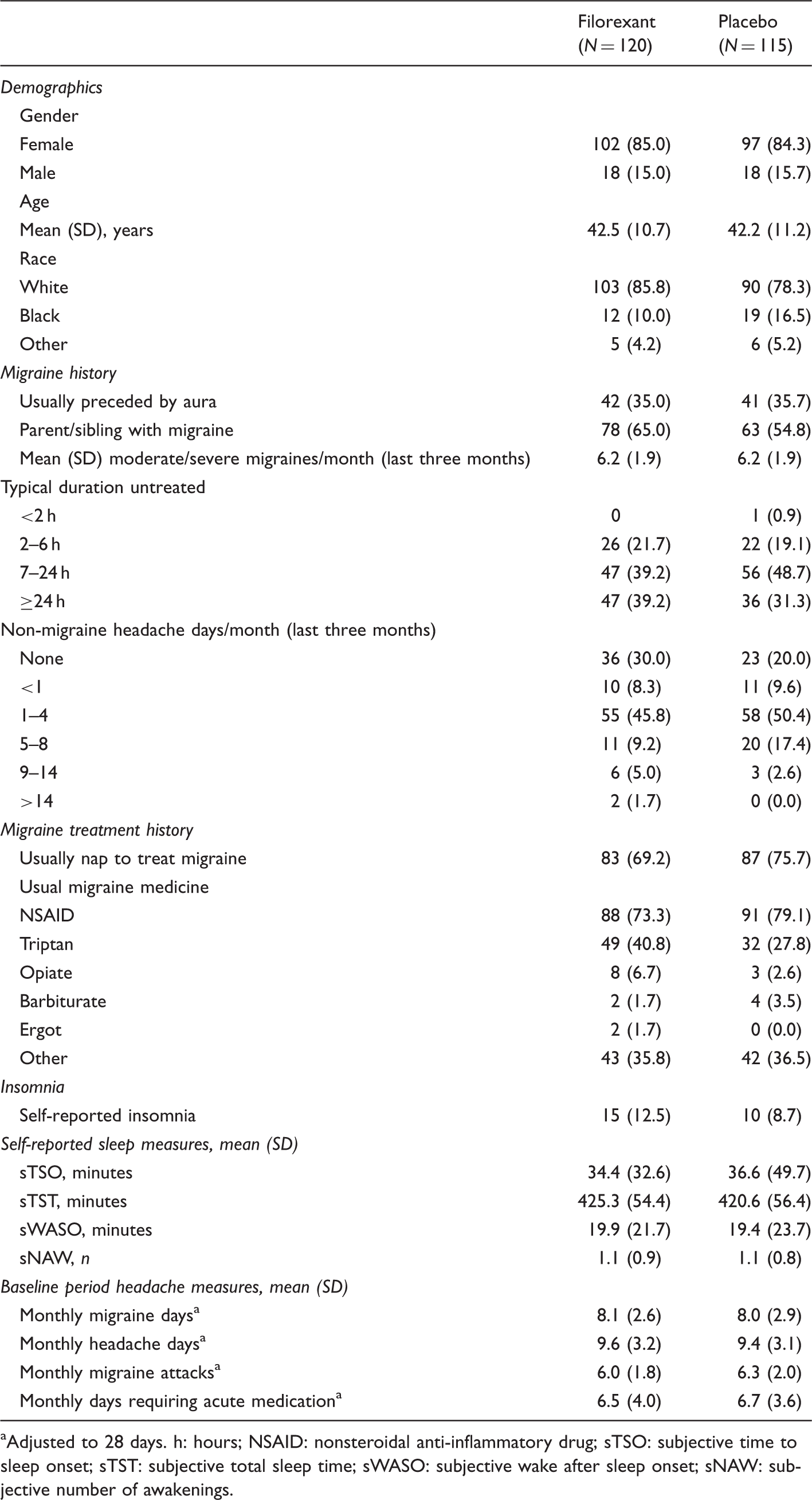
aAdjusted to 28 days. h: hours; NSAID: nonsteroidal anti-inflammatory drug; sTSO: subjective time to sleep onset; sTST: subjective total sleep time; sWASO: subjective wake after sleep onset; sNAW: subjective number of awakenings.
The average number of moderate or severe migraines per month over the three months prior to screening was six. During the four-week baseline period, the mean number of monthly migraine days was eight. With regard to sleep characteristics, 72% reported that they usually napped to treat migraine, and 11% had self-reported insomnia.
Efficacy
Change from baseline on outcome measures averaged over the three-month treatment period (migraine and headache measures) or at Month 3 (sleep measures).

Monthly rates were adjusted to 28 days. aFor responder analyses, N = 115 for filorexant and N = 112 for placebo; for sleep analyses, N = 94 for filorexant and 97 for placebo. sTSO: subjective time to sleep onset; sTST: subjective total sleep time; sWASO: subjective wake after sleep onset; sNAW: subjective number of awakenings; LS: least squares; SE: standard error; CI: confidence interval; OR: odds ratio. Analyses should be considered descriptive and exploratory because the primary endpoint did not achieve statistical significance.
General tolerability
Summary of adverse events during the three-month treatment period: number (%) of patients.
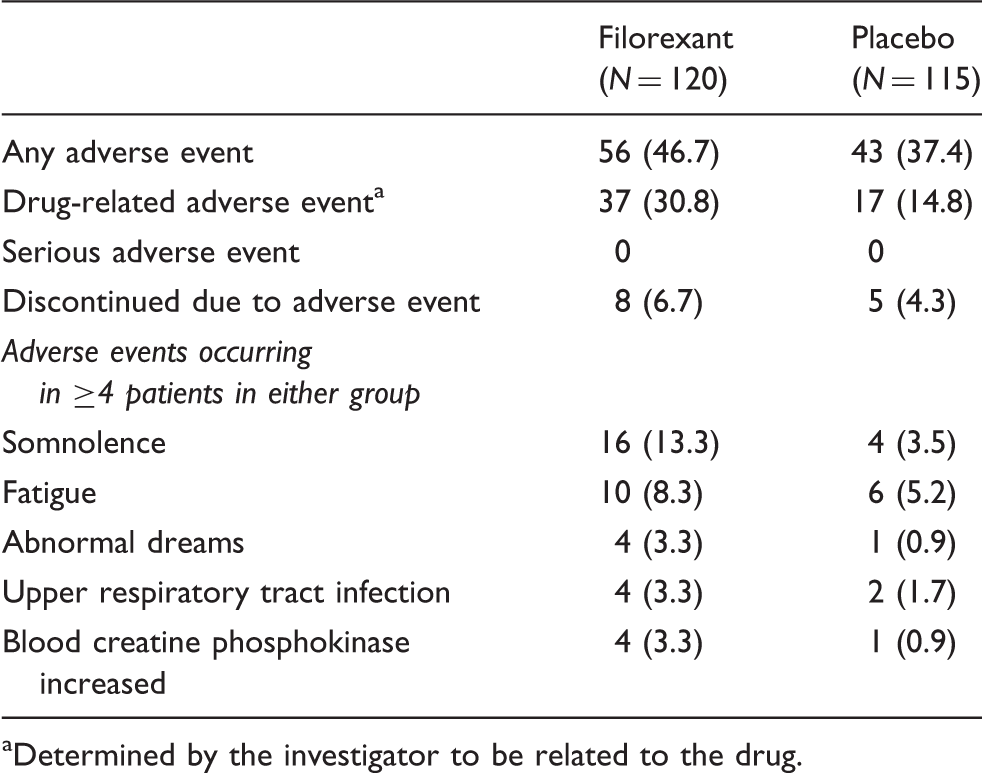
aDetermined by the investigator to be related to the drug.
During the two-week double-blind run-out period that followed the primary three-month treatment period, the number of patients reporting any adverse events were five of 50 (10.0%) in those previously on filorexant remaining on filorexant, eight of 47 (17.0%) in those previously on filorexant switched to placebo, and eight of 101 (7.9%) in those previously on placebo remaining on placebo. There were no ECIs and no discontinuations due to adverse events during the two-week run-out.
No clinically relevant mean changes in laboratory or ECG parameters or vital signs were observed during the study.
Discussion
The findings from this study of 235 patients treated with filorexant or placebo do not provide evidence that orexin receptor antagonism is an effective mechanism for preventing migraine.
Several factors limit the interpretation of these results. The study was powered to detect a difference between treatments of 1.6 days per month, and we cannot rule out the possibility that a smaller effect might have been observed in a larger sample. We also note that the majority of the sample did not experience sleep problems, and it is possible that orexin receptor antagonism could be effective in a subgroup of patients for whom migraine is associated with sleep disturbance. While there appeared to be a larger effect in patients with sleep disturbance at baseline, this observation should be treated very cautiously given the small sample of patients in whom it was observed.
Perhaps the most important limitation of this study is the possibility that study-specific issues precluded the observation of a therapeutic effect that the mechanism could in fact have provided. In the absence of a positive control, we cannot definitively exclude this possibility. However, the weight of evidence suggests that this is not the case, and that the study findings correctly represent a negative rather than a “failed” study. In particular we note that the overall characteristics of the study population are similar to those in other prophylaxis studies (25,27) and that the degree of non-specific (“placebo”) response was limited.
It is important to consider whether the pharmacological profile of filorexant and the particular dose evaluated were adequate to furnish a test of the orexin receptor antagonism mechanism for migraine prophylaxis. A 10-mg dose was selected for evaluation because this dose was previously shown to be effective for treating insomnia (21,22) and therefore has known central activity. The central activity of the 10-mg dose was confirmed by the increase in central nervous system (CNS) adverse events in the present study. Given that migraine patients are presumed to have an easily “excited” nervous system, it is possible that the 10-mg dose was sub-therapeutic for most migraine patients, and we cannot exclude the possibility that a higher dose would have been effective for preventing migraine.
Another aspect of filorexant’s pharmacological profile that could have affected the observed outcome is related to the need for nighttime dosing in order to avoid debilitating daytime sedation. The relatively rapid Tmax (∼2 hours) and short half-life (three to five hours) of filorexant means drug exposure during the day was minimal. If efficacy in migraine requires higher drug exposure at the time of the migraine onset, then this could account for the failure to observe an effect. However, because dosing an orexin receptor antagonist during the day may lead to sedation, if daytime dosing is required it is unlikely that orexin receptor antagonism could be a viable treatment for most patients.
Filorexant is a dual orexin receptor antagonist, with activity at both orexin (Ox)1 and Ox2 receptors. Relatively little is known about the respective roles of the specific receptor subtypes in migraine or pain, but some preclinical data suggest that Ox1 and Ox2 receptor activation may have differential effects. In particular, Ox1 receptor activation appears to have an analgesic effect via decrease of A- and C–fiber response to dural stimulation while Ox2 receptor activation appears to have a pronociceptive effect via increase of A- and C-fiber response to dural stimulation (15,16). It is conceivable, albeit unlikely, that selective orexin receptor antagonists, with activity at only the Ox1 receptor or only the Ox2 receptor, could exhibit analgesic effects in migraine patients.
With regard to safety, the present findings are in line with expectations based on previous studies of the orexin receptor antagonists almorexant and suvorexant in healthy individuals and insomnia patients (8,9,28,29). Overall, treatment was generally well tolerated. There were no serious adverse events and no significant safety concerns emerged over three months of nightly use in migraine patients or following the abrupt discontinuation of filorexant. Somnolence was the most common adverse event associated with filorexant but resulted in study discontinuation in only a small number of patients.
Clinical implications
Nighttime administration of filorexant at a dose associated with clear sleep-promoting effects in a previous study of insomnia patients was not effective in the prevention of migraine. These results do not support the efficacy of orexin receptor antagonism with filorexant as a potential therapeutic approach in migraine.
Footnotes
Funding
This work was supported by Merck & Co. Inc, Whitehouse Station, NJ. The funding organization was involved in the design and conduct of the study, the collection, management, analysis, and interpretation of the data, and the preparation, review, and approval of the manuscript.
Conflicts of interest
AC, YZ, SJ, CL, WJH, KMC and DM are current or former employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co. Inc, Whitehouse Station, NJ, and own or owned stock/stock options in Merck.
RC currently serves on several advisory boards: Allergan, Boston Scientific, MAP Pharmaceuticals, Merck & Co. Inc, Nautilus Neuroscience, Novartis, NuPathe, Optinose, Pfizer, and Zogenix. He also receives research grants from Allergan, Amgen, Arteaus, AstraZeneca, Boston Scientific, Bristol-Myers Squibb, ElectroCore, EpiQ, Forest Pharmaceuticals, Fortis Spectrum, GlaxoSmithKline, Janssen Services, MAP Pharmaceuticals, Merck & Co Inc, Nico Worldwide, NuPathe, OptiNose, Pearl Therapeutics, PuraMed Bioscience, Tian Medical, Transcept, and Zogenix. Dr Cady has provided consulting services for Allergan, Amgen, Avanir, Evidera Inc, Merck & Co. Inc, Pfizer, Transcept, and Zogenix.
Acknowledgments
The authors would like to acknowledge Sheila Erespe of Merck for her submission assistance. The following investigators participated in the study: Eugene Andruczyk, Philadelphia, PA; Elizabeth Bretton, Albuquerque, NM; Roger K. Cady, Springfield, MO; Mahan Chehrenama, McLean, VA; James R. Clark, Charlottesville, VA; Steven Folkerth, Las Vegas, NV; Martin K. Kankam, Overland Park, KS; William D. Koltun, San Diego, CA; David B. Kudrow, Santa Monica, CA; Mark Eliot Kutner, Miami, FL; Lisa K. Mannix, West Chester, OH; Vicki E. Miller, Houston, TX; Antoinette Alexia Pragalos, Cincinnati, OH; Jay Jonas Rubin, Ocala, FL; Joel R. Saper, Ann Arbor, MI; Cynthia B Strout, Mount Pleasant, SC; Albert Q. Tejada, Scottsdale, AZ; Michael M. Tuchman, Palm Beach Gardens, FL; Scott A. Wilson, Cumberland, RI.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.