
Abstract
Methods: Four hundred and sixty-one adult subjects with migraine were randomised to one of five treatments, the oral antagonist at the calcitonin gene-related peptide (CGRP) receptor BI 44370 TA (50 mg, 200 mg, 400 mg), active comparator eletriptan 40 mg or placebo. The analysis included 341 subjects who took study medication.
Results: The primary endpoint, pain-free after two hours, was reached by significantly more subjects in the BI 44370 TA 400 mg (20/73 = 27.4%) and eletriptan 40 mg (24/69 = 34.8%) groups compared to placebo (6/70 = 8.6%, p = .0016), but not by subjects in the BI 44370 TA 200 mg group (14/65 = 21.5%). The effect of 50 mg BI 44370 TA (5/64 = 7.8%) was similar to that of placebo. Analysis of secondary endpoints supported the conclusion from the primary analysis. The frequency of adverse events was low in all groups.
Conclusion: Efficacy of BI 44370 TA was shown in a dose-dependent manner in the treatment of acute migraine attacks.
Introduction
Migraine is a condition of major public health concern, as approximately 12% of the population (with higher prevalence in women than in men) suffer from recurrent migraine attacks that interfere with their daily lives (1,2). Symptomatic treatment of migraine can be achieved with 5-hydroxytryptamin1B/1D-receptor agonists (triptans), ergot alkaloids, nonsteroidal anti-inflammatory drugs (NSAIDs) and with combination of analgesics and antiemetics. However, migraine sufferers perceive a wide range of treatment needs that are not fully met by currently available therapies. These include a faster onset of action, a lower incidence of drug-induced side effects and an improvement of response rate (usually 60–70%) and relapse rate (up to 40% of initial responders suffer from recurrent headaches within several hours) (3,4). This emphasises the need for novel therapies with improved efficacy and good tolerability without the perceived drawback of cardiovascular liability of the triptans (5).
Acute migraine is a neurovascular disease thought to be associated with dilation of cranial vessels and activation of the trigemino-vascular system (6–9). Stimulation of the trigeminal ganglion in humans leads to release of calcitonin gene-related peptide (CGRP) from perivascular sensory axons to mediate vasodilation of mainly extracerebral intracranial arteries and arteriovenous anastomoses (10). Recently, it has been demonstrated that CGRP is also involved in pain transmission within the trigeminal nerve system in the central nervous system (CNS) (11). The inhibition of CGRP-mediated effects, possibly at both sites, attenuates migraine symptoms.
BI 44370 TA is an orally available CGRP antagonist that showed good tolerability and very low incidence of adverse events (AEs) in phase I trials. Clinical trials examining other CGRP antagonists for the treatment of acute migraine support the role of CGRP in migraine pathogenesis and showed effective treatment of migraine attacks (12–15).
Therefore, treatment with CGRP antagonists may offer advantages over the existing therapies, in particular in subjects with contraindications for triptans or subjects who cannot tolerate triptans due to adverse events. The objective of this trial was to assess the safety, tolerability and efficacy of three doses of BI 44370 TA (50 mg, 200 mg, 400 mg) in the treatment of an acute migraine attack of moderate or severe intensity compared to placebo.
Methods
Subjects
The study (ClinicalTrials.gov NCT00751803) was approved by ethical committees of each participating headache centre. All subjects provided written informed consent in accordance with the International Conference on Harmonisation—Good Clinical Practice (ICH-GCP) and local legislation prior to any trial specific procedure. Male and female subjects were eligible for the study if they were ≥18 and ≤65 years of age, had a history of migraine with or without aura (International Classification of Headache Disorders, 2nd edition) (16), for at least one year and were in general good health. In the three months prior to the screening visit, subjects had to have two to eight migraine attacks of moderate to severe intensity per month, each lasting at least six hours. Subjects were excluded if they fulfilled contraindications listed in the eletriptan summary of product characteristics, suffered from other pain syndromes, were taking migraine prevention medication or other pain medication on more than 10 days per month, were classified as treatment resistant by the investigator or had signs of liver injury (history of severe hepatic disease or elevated liver function test >2x upper limit of normal [ULN]). Subjects on benzodiazepines, antidepressants or potent CYP3A4 inhibitors (cytochrome P450) were not allowed to participate in the trial.
The study was conducted at 47 sites in Europe from August 2008 to May 2009, inclusive.
Study design
This was a randomised, double-blind, double-dummy, placebo- and active-controlled, parallel-group, outpatient study in subjects with an acute migraine attack. Subjects were allocated to one of the five treatment groups in equal ratios. Three doses of BI 44370 TA were tested: 50 mg, 200 mg and 400 mg. On the basis of pharmacokinetic studies, it was estimated that the 200 mg dose yields concentrations targeted for efficacy plateau. The 50 mg and 400 mg doses were added to show dose-response relationship. The active comparator used was eletriptan (Relpax®) at the dose of 40 mg. The fifth arm was placebo.
BI 44370 TA was supplied as tablets of 50 mg and 200 mg strength. Eletriptan was over-capsulated to keep the study blind. Matching placebos were available. Subjects were allocated to treatment according to a computer-generated randomisation list, created by the internal Clinical Trial Support group, using a block size of 10 subjects. The treatment assignment was carried out by a third-party central randomisation service (IXRS, ALMAC Clinical Technologies, USA) stratified by centre. Persons involved in the conduct and analysis of the trial had no access to the treatment allocation prior to database lock.
Procedures
Subjects who met all the study entry criteria were randomised and provided with study drug to be taken on an outpatient basis. They were instructed to take study medication to treat their next migraine attack when the migraine pain was moderate or severe, and when no additional requirements were violated (i.e. a treatable migraine attack). Subjects with exclusively non-treatable migraine attacks within 60 days after randomisation or who did not take study medication for any other reason were considered dropouts. Migraine attacks were defined as non-treatable when they occurred in the middle of the night or when another pain medication was taken within 48 hours preceding the attack. Rescue medication, with the exception of triptans, ergotamines or domperidone, was allowed beginning two hours after intake of study medication to treat persistent or recurrent migraine attack.
During the 48 hours after the intake of study medication, subjective evaluation of the symptoms and use of rescue medication was recorded by the subject in a paper diary at pre-specified time intervals. Subjects were instructed to return to the study site within three to seven days after the migraine attack for the final visit. At this visit, the investigator reviewed the diary, assessed medication compliance, and performed monitoring of tolerability and safety (adverse events, physical examination including vital signs, laboratory test and electrocardiogram [ECG]).
Headache severity was recorded using a four-grade scale (no pain, mild pain, moderate pain, severe pain) at baseline (0 hours—time of taking study medication) and at 0.5, 1, 1.5, 2, 24 and 48 hours after intake of study medication. Presence or absence of associated symptoms (nausea, vomiting, photophobia, phonophobia) and rating of functional disability (4-grade scale: normal, mildly impaired, moderately impaired, severely impaired) were recorded at the same time points as headache severity.
Statistical analysis
The primary endpoint was pain-free response (defined as reduction of severe or moderate headache to no headache) two hours after dosing. The primary comparison was between BI 44370 TA doses and placebo; one-sided p values of at most .025 were considered as significant. Type I error rate was preserved by a fixed sequence testing procedure starting with the highest BI 44370 TA dose, eletriptan was included for benchmarking only.
Protocol defined secondary efficacy endpoints included pain relief (reduction of severe or moderate to mild or no headache), sustained pain-free and sustained pain relief (defined as pain-free or pain relief at two hours without relapse up to 24 and 48 hours, respectively), relapse, absence of migraine symptoms (nausea, vomiting, photophobia, phonophobia), functional disability, time to meaningful relief, global evaluation of study medication and use of rescue medication. Additional post-hoc analysis were performed for the following efficacy endpoints: total migraine-free (no pain and absence of migraine symptoms); sustained pain-free response and no AEs (SNAE) up to 24 and 48 hours; and improvement in functional disability score (total improvement: from 2–3 to 0; “relief”: from 2–3 to 0–1). Secondary safety endpoints included incidence of adverse events and laboratory parameters.
Responder rates were statistically compared based on odds ratios. Exact one-sided p values were calculated with PROC STRATIFY (exact conditional inference stratified by country—no mid-p-correction). Time to event endpoints was illustrated using Kaplan Meier curves, and these were compared using log rank test.
All treated subjects who had at least one post-dose efficacy assessment were included in the efficacy analysis (full analysis set [FAS]).
Missing data post-baseline were imputed using a last-observation-carried-forward approach. For time-to-event variables, missing time data were set to the worst possible criterion (e.g. time to meaningful relief >two hours). For subjects who required rescue medication, all efficacy assessments following intake of rescue medication were analysed in the least favourable category (e.g. headache severity = severe). The same principle was applied to subjects with protocol violations.
All subjects who took study treatment were included in the safety assessment. Adverse events occurring within 48 hours after dosing were assigned to treatment.
On the basis of a sample size of 69 subjects per treatment group, expecting a placebo response rate of about 10% and an active response rate of about 30%, and using a one-sided significance level of .025, the study had at least 80% power to show superiority of the highest-dose group over placebo on the primary endpoint. To accommodate for a dropout rate of almost 15%, the total sample size was set to 410 subjects (82 per group).
Results
The trial profile is shown in the Figure 1. A total of 341 subjects were treated with study medication: 70 subjects with placebo, 64 subjects with 50 mg BI 44370 TA, 65 subjects with 200 mg BI 44370 TA, 73 subjects with 400 mg BI 44370 TA and 69 subjects with 40 mg eletriptan. The planned number of 69 subjects per treatment group for statistical analyses was not reached in the 50 mg and 200 mg BI 44370 TA groups. The imbalance of treated subjects per treatment group occurred by chance.
Trial profile. Values indicate number of subjects. PV = protocol violation. AE = adverse event. TS = treated set. FAS = full analysis set. PPS = per protocol set.
Demographics and baseline characteristics
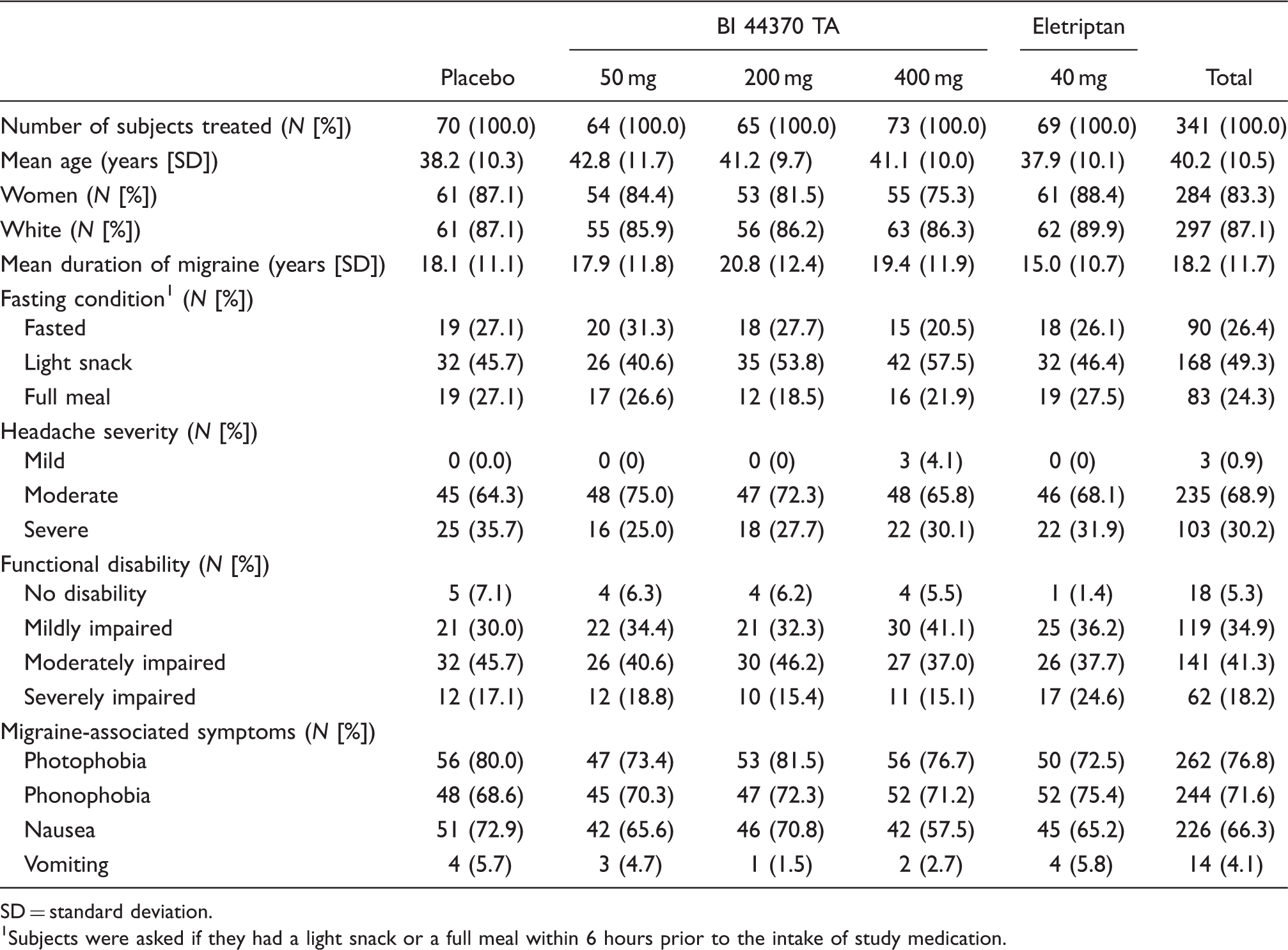
SD = standard deviation.
Subjects were asked if they had a light snack or a full meal within 6 hours prior to the intake of study medication.
Efficacy outcomes are summarised in Table 2. The 400 mg doses of BI 44370 TA and eletriptan 40 mg were more effective than placebo on the primary endpoint pain-free at two hours (statistically significant; Figure 2). Subjects treated with the 200 mg BI 44370 TA dose showed a higher pain-free response at two hours than placebo; however, the effect did not reach statistical significance (p > .025, one-sided significance level). The 50 mg dose of BI 44370 TA had an efficacy similar to that of placebo.
Pain-free response at the pre-selected time points. Percentage of subjects reporting no headache, without use of rescue medication, after treatment with placebo (white bar), BI 44370 TA (grey bars) and eletriptan (black bar); exact 95% confidence intervals by Clopper and Pearson included as error bars. Level of significance: *p < .025, **p < .005, ***p < .0005 for the active vs. placebo pairwise comparison. hh = hours. mm = minutes. Efficacy endpoints h= hours. CI = confidence interval. Exact conditional inference stratified by country. Starting 2 hours after intake of study medication, subjects were allowed to take rescue medication, if needed. The sustained pain-free and sustained pain relief results include only subjects who did not take any rescue medication. Level of significance: *p < .025, **p < .005, ***p < .0005 for the active vs. placebo pairwise comparison.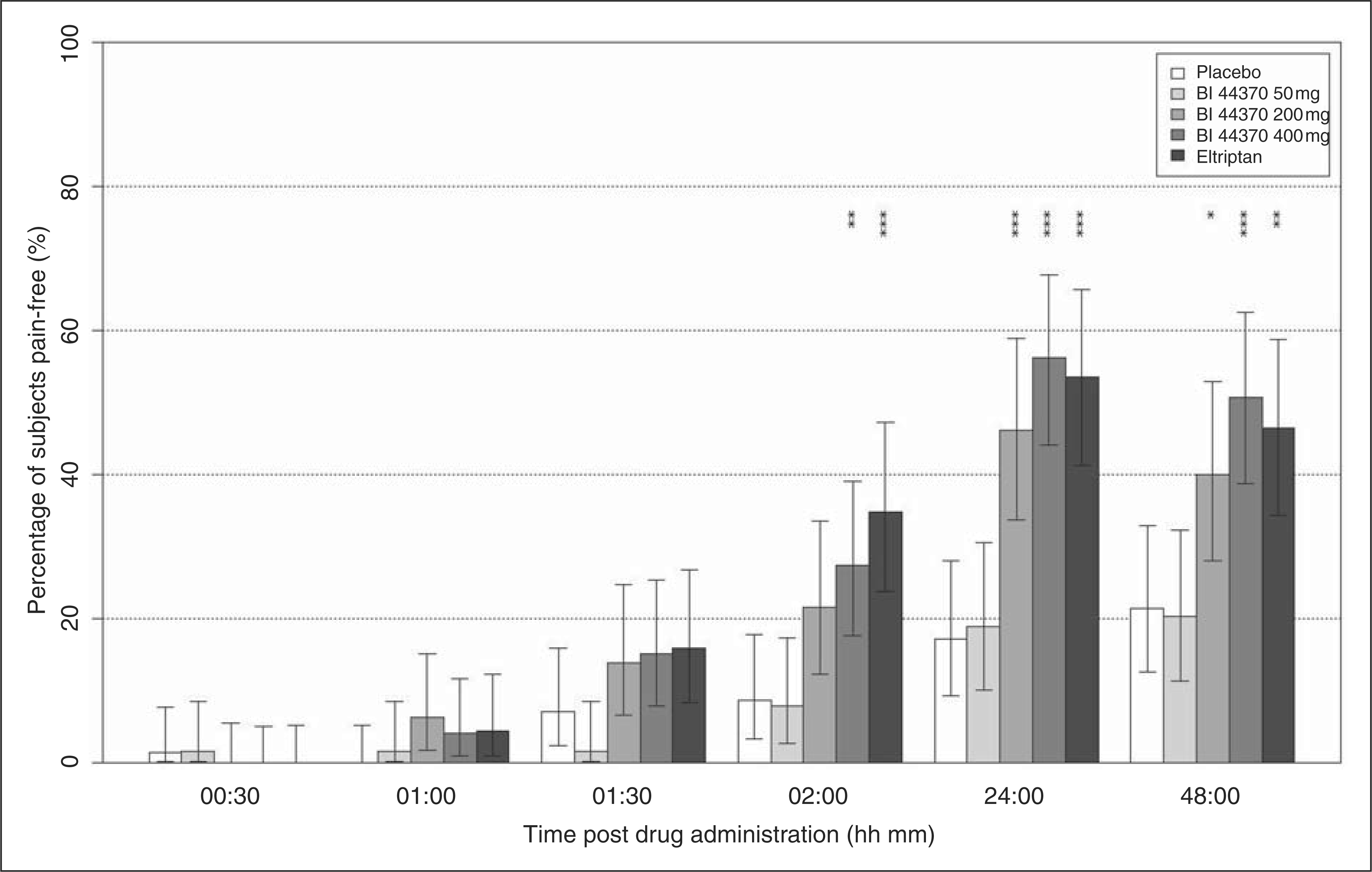

A similar dose-dependent pattern of response was found for a number of secondary endpoints, such as pain relief, absence of photophobia, phonophobia and nausea, as well as post-hoc secondary endpoints such as total migraine-free and improvement in the functional disability scores (Table 2). The incidence of vomiting at baseline was too low (Table 1) to allow for any improvement following treatment. The analyses of sustained pain-free and sustained pain relief indicate a favourable duration of efficacy of BI 44370 TA (Table 2). In accordance with these results, subjects treated with BI 44370 TA at doses of 200 and 400 mg, as well as those treated with eletriptan 40 mg, needed less rescue medication than subjects on placebo or on 50 mg of BI 44370 TA (Table 3). The 400 mg dose of BI 44370 TA was also significantly better over placebo on the combined post-hoc endpoint SNAE (Figure 3).
Sustained pain-free and no adverse event (SNAE) was a post-hoc secondary outcome measure and was calculated as a percentage of subjects pain-free from 2 to 24 hours, and from 2 to 48 hours, furthermore reporting no adverse events during the respective period; exact 95% confidence intervals by Clopper and Pearson included as error bars. Level of significance: *p < .025 for the active vs. placebo pairwise comparison. h = hours. Subjects using rescue medication h= hours. Level of significance: *p < .025, **p < .005, ***p < .0005 for the active vs. placebo pairwise comparison.

Effects of study medication on functional disability were assessed as percentage of subjects with no, mild, moderate or severe disability at each pre-defined time point. Results showed that at two hours post-treatment, 33% of subjects treated with placebo reported severe disability whereas no disability was reported by subjects treated with BI 44370 TA 200 mg (31%), 400 mg (34%) and eletriptan (34%). The post-hoc assessments focused on evaluating improvement in functional disability scores (i.e. in relation to baseline). Improvement in the functional disability score also showed a dose-response pattern. Subjects treated with the 400 mg dose and eletriptan had the most robust improvement in the functional disability score. This pattern was observed for any improvement in the score as well as for improvement from 2–3 score to 0 score (total improvement; Table 2) or from 2–3 score to 0–1 score (“relief”; Table 2). The first sign of efficacy was pain relief observed one hour after intake of 400 mg of BI 44370 TA (32.9%, p = .0093). At the time points of 24 and 48 hours after intake of study drug (without any rescue medication), BI 44370 TA at 200 and 400 mg and eletriptan 40 mg showed a robust effect on both pain relief and pain-free response (Figure 2).
Relapse was defined as re-appearance of pain after pain-free response at two hours. A comparison of relapse rates is meaningful only for groups with similar and sufficiently high responder rates. In this study, groups treated with placebo and 50 mg BI 44370 TA did not have a sufficient number of responders to evaluate relapse. Therefore, only the three effective groups (BI 44370 TA 200 and 400 mg and eletriptan 40 mg) were compared descriptively. We observed a higher incidence of relapse in the eletriptan group when compared to the groups treated with BI 44370 TA (200 mg BI 44370 TA: 2/14 responders [14%]; 400 mg BI 44370 TA: 4/20 [20%]; eletriptan 40 mg: 11/24 [46%]).
Global evaluation of medication was recorded 48 hours after treatment on a seven-point categorical scale ranging from excellent to extremely poor. The distribution over categories of active groups was compared to the distribution in the placebo group. Subjects treated with 200 mg and 400 mg of BI 44370 TA and with eletriptan 40 mg had different distribution from placebo, whereas subjects treated with 50 mg BI 44370 TA had distribution similar to that of placebo (Figure 4). For example, 77% of subjects treated with placebo and 78% subjects treated with 50 mg BI 44370 TA evaluated the study medication as extremely poor but only 49% treated with 200 mg, 40% with 400 mg BI 44370 TA and 44% with eletriptan.
Global evaluation of study medication by subject. Percentage of subjects evaluating study medication as excellent, very good, good, neither good nor bad, poor, very poor or extremely poor. Cochran-Mantel-Haenzel row mean score test was applied to compare the distribution over categories within treatment groups. Groups treated with BI 44370 TA 200 mg and 400 mg and with eletriptan 40 mg were significantly different from placebo.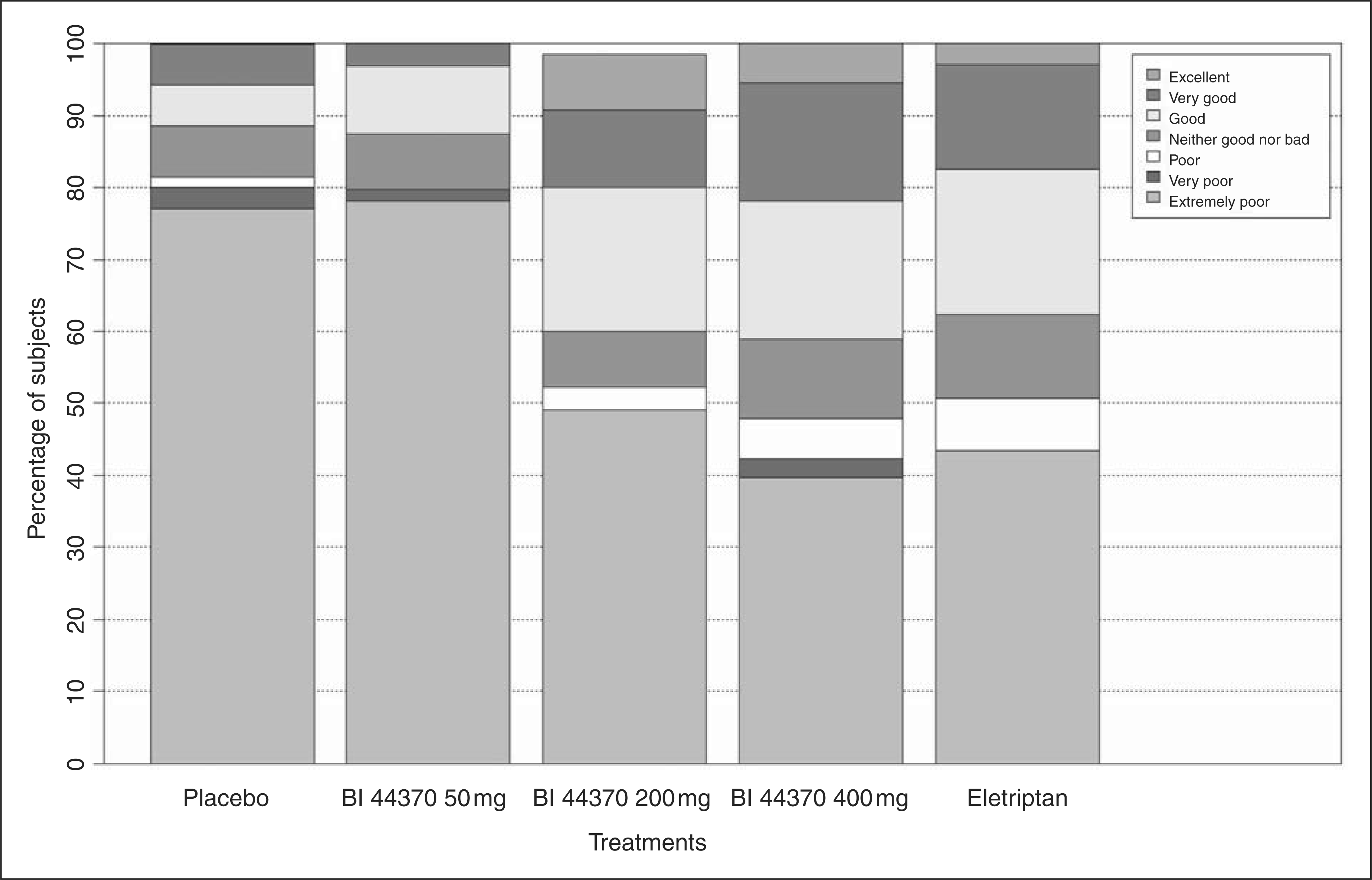
Data on the time to meaningful relief were collected. However, a large number of subjects did not report meaningful relief in any group within two hours after intake of study medication. Therefore, medians could not be determined. The same is true for time to rescue medication. Only in the placebo and 50 mg BI 44370 TA groups were there enough subjects who took rescue medication, and therefore, median could be computed (6′12 minutes for placebo, 7'25 minutes for 50 mg BI 44370 TA). Kaplan Meier curves of active groups compared to placebo showed that 50 mg BI 44370 TA coincide to placebo while 200 mg and 400 mg BI 44370 TA and eletriptan 40 mg differ from placebo for both outcomes.
Incidence of adverse events during the treatment period (intake of study medication + 48 hours)
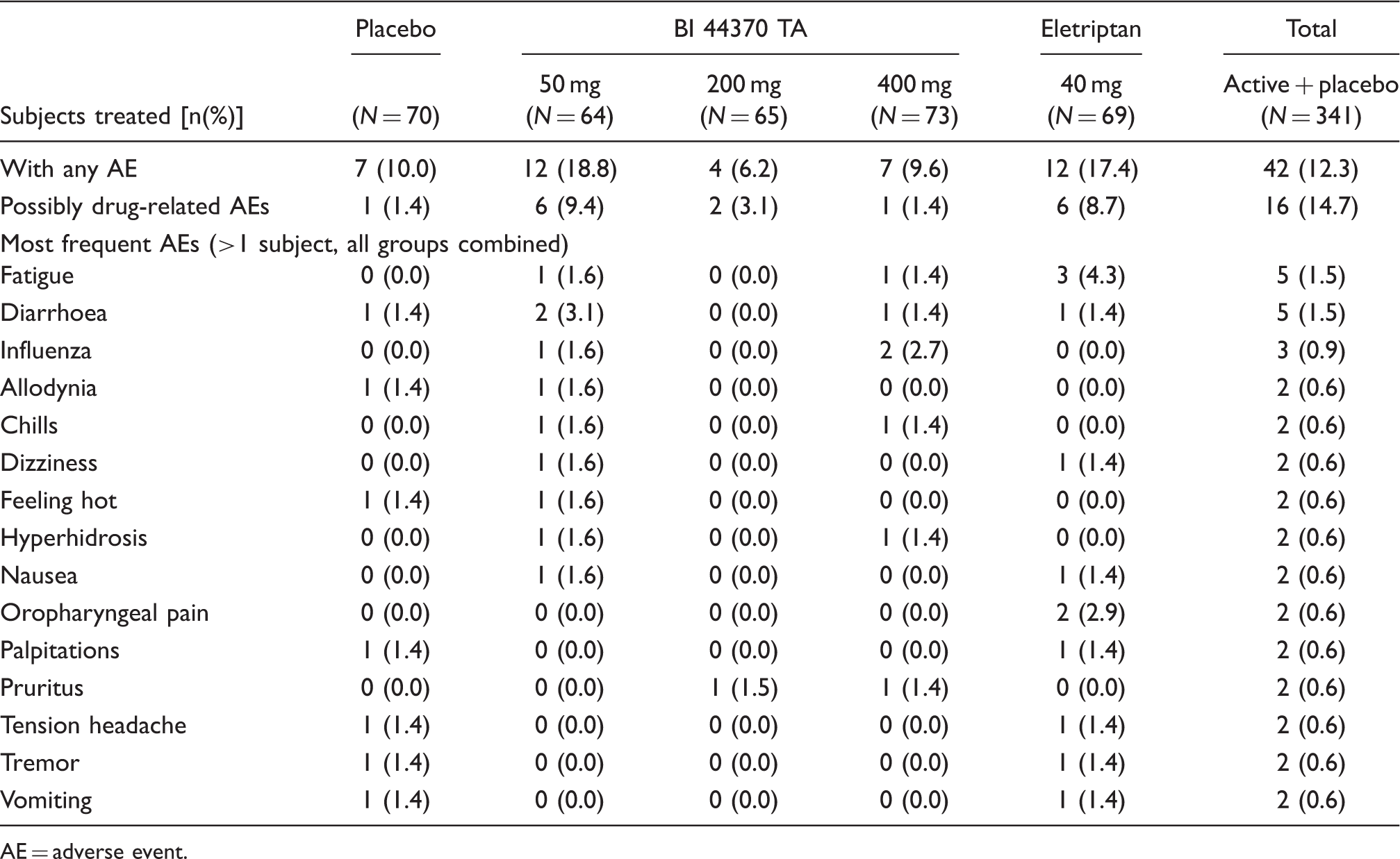
AE = adverse event.
Abnormalities of laboratory values were rare, and there were no clinically relevant differences between treatment groups. One subject had elevated values of alanine transaminase (ALT > 5x upper limit normal (ULN)), aspartate transfaminase (AST > 3x ULN), and gamma glutamyl transpeptidase (GGT > 5x ULN) but no bilirubin elevation, five days after a single administration of 400 mg BI 44370 TA. At baseline, only a slight increase of ALT (<2x ULN) was found. Serology screen for hepatitis was negative. Detailed review of the subject’s medical history revealed a daily use of amitriptyline for depression since 1998, propranolol for migraine since 2007 and an intermittent use of analgesics at high doses. Inclusion was classified as a protocol violation as exclusion criteria were met (e.g. clinically significant psychiatric disorder, migraine preventive treatment). Moreover, this subject had influenza and took Afebryl® (ASA + paracetamol) during the time of the migraine attack, even though intake of study medication was not allowed within 48 hours from intake of pain medication. During the follow-up examinations, cholecystitis was detected. The values returned to baseline within 14 days. No other events of liver function abnormalities were observed in this study.
Discussion
This proof-of-concept and dose-finding study assessed efficacy and tolerability of three doses of BI 44370 TA (50 mg, 200 mg and 400 mg) in adult migraine subjects with headache pain of moderate to severe intensity, in comparison with placebo. An active comparator (40 mg eletriptan) was included for assay sensitivity. Efficacy in the treatment of acute migraine attacks (i.e. proof-of-concept) was obtained for BI 44370 TA. Dose-response relationship was observed: 400 mg ≥ 200 mg >50 mg ≈ placebo. The orally administered CGRP antagonist BI 44370 TA was effective in the treatment of migraine with pain of moderate or severe intensity. The 400 mg dose of BI 44370 TA was significantly better than placebo at all efficacy endpoints, similarly to eletriptan 40 mg. The 200 mg BI 44370 TA was superior to placebo even though it failed to reach statistical significance for some of the endpoints (e.g. pain-free at two hours, sustained pain-free). The placebo pain-free response was 8.6% and was in the range published previously for other migraine trials (17,18).
In the 50 mg and 200 mg BI 44370 TA groups, the planned number of 69 subjects per treatment group (which were required to detect statistically significant differences between treatments with 80% power) was not reached. This small decrease in subject numbers per treatment group possibly could have influenced the results of this trial.
The main strength of the present study is the placebo-controlled design with an active comparator. Another strength is the use of three different doses of BI 44370 TA, which allowed identifying a non-effective dose (50 mg) and showing dose response. A potential shortcoming could be the use of five treatment groups. Subjects might expect to receive active drug in a trial with a 4 : 1 randomisation ratio, which could increase the placebo response. However, the placebo rate in this trial was in the range of reported placebo response in other migraine trials with oral medications (17,18). Generalisability of the trial results should only be considered in the context of the present inclusion and exclusion criteria. A final evaluation of the benefit of BI 43370TA will only be possible once the phase III program has been undertaken.
It should be noted that subjects treated with 400 mg of BI 44370 TA showed similar pain-free, pain relief and total migraine-free responses as subjects treated with other CGRP antagonists (19,20). Results observed in the eletriptan 40 mg group were comparable to those found in previous eletriptan trials (21,22).
Relapse is an acknowledged problem of triptans, and often leads to dissatisfaction (23,24) and discontinuation of treatment (25,26). Recently, the value of the relapse rate as an endpoint has been questioned (27), as the incidence of relapse is highly dependent on the initial response rate, as mentioned above. Sustained pain-free and sustained pain relief have been proposed instead as more reliable and statistically more correct endpoints. Data on sustained pain-free and sustained pain relief responses revealed that a significantly higher percentage of subjects treated with 400 mg BI 44370 TA remained pain-free up to 48 hours if compared to placebo (Table 2). This indicates a favourable duration of efficacy of BI 44370 TA.
Blinding was achieved by encapsulation of eletriptan. In vitro comparisons of dissolution profiles of eletriptan tablets (40 mg) and over-capsulated eletriptan tablets (40 mg) at the three pH = 1, 4.5 and 6.8 were found similar as both products dissolved ≥85% of the label amount of the drug in <15 minutes (internal data). In vivo comparison of bioavailability was not performed. It was previously reported that encapsulation of sumatriptan delays absorption for up to two hours after intake, particularly during migraine (28). However, differences in efficacy of sumatriptan were found in clinical trials using encapsulated sumatriptan as active comparator (29–31). Efficacy of eletriptan in our study was similar to efficacy published previously (22,32). Even though our results are not supportive of the idea that encapsulation might have compromised the efficacy of eletriptan, it cannot be excluded.
BI 44370 TA was safe and well tolerated. Incidence of AEs was low and similar between groups. No AE occurred with frequency of 5% or more. No clinically relevant changes in vital signs or ECG were detected across the studies performed. This supports the view that CGRP antagonists are well tolerated, as previously reported for olcegepant (33) and telcagepant (14,34). Increased liver function tests were found in one subject five days after administration of a single dose of 400 mg BI 44370 TA. Detailed review of the subject’s medical status showed concomitant medications and diseases as well as alcohol consume. It is difficult to draw a conclusion, therefore, as the elevation could be due to our compound, an alcohol intake (supported by simultaneous increase of GGT), increased susceptibility of the liver by co-medications and subject’s health condition or any combination of the above. Moreover, no other liver function test abnormalities were found in our clinical studies with this compound (internal data).
One potential benefit of CGRP receptor antagonists for treatment of acute migraine is the alleged lack of vasoconstriction. This may allow for a safe administration of this class of compounds in migraine subjects with cardiovascular disease. However, such subjects were excluded from the study with BI 44370 TA because of the contraindications for eletriptan.
Conclusion
In conclusion, proof-of-concept was shown in a dose-dependent manner for BI 44370 TA in the treatment of acute migraine attacks. Our findings support and extend the previous proof-of-concept findings with CGRP antagonists olcegepant (35) and telcagepant (14,36). The beneficial effect of BI 44370 TA in subjects with an acute migraine attack has to be replicated in a large phase III study.
Disclosures
Prof Dr Hans-Christoph Diener has received honoraria for participation in clinical trials, contribution to advisory boards or oral presentations from Addex Pharma, Allergan, Almirall, AstraZeneca, Bayer Vital, Berlin Chemie, Coherex Medical, CoLucid, Boehringer Ingelheim, Bristol-Myers Squibb, GlaxoSmithKline (GSK), Grünenthal, Janssen-Cilag, Lilly, Hoffman La Roche, 3 M Medica, Minster, MSD, Novartis, Johnson & Johnson, Pierre Fabre, Pfizer, Schaper and Brümmer, Sanofi Aventis andWeber & Weber. Financial support for research projects was provided by Allergan, Almirall, AstraZeneca, Bayer, GSK, Janssen-Cilag and Pfizer. Headache research at the Department of Neurology in Essen is supported by the German Research Council (DFG), the German Ministry of Education and Research (BMBF) and the European Union. H.C. Diener has no ownership interest and does not own stocks of any pharmaceutical company.
Prof Piero Barbanti has received honoraria for participation in clinical trials, contribution in advisory boards or oral presentations by Merck Sharp & Dohme, Boehringer Ingelheim, GSK, Lusofarmaco and Janssen Cilag. P. Barbanti has no ownership interest of any pharmaceutical company.
Prof Dr Carl Dahlöf is or has been a consultant/scientific advisor on advisory boards, clinical trials, investigator-initiated trials and/or speaker for Allergan, Almirall Prodesfarma, AstraZeneca, Bayer, Bristol-Myers Squibb, Coherex, Eisai, GSK, Janssen-Cilag, Johnson & Johnson, Merck, Lilly, Lundbeck, NMT Medical, Novartis, Nycomed, Ortho-McNeil Pharmaceutical, Pharmacia, Pharmnovo, Pfizer, Pierre Fabre St. Jude Medical, Takeda and Vernalis. C. Dahlöf has no ownership interest in and does not own stocks of any pharmaceutical company.
Dr. Uwe Reuter has received honoraria for participation in clinical trials, contribution to advisory boards or oral presentations from Addex Pharma, Allergan, Almirall, AstraZeneca, Berlin Chemie, CoLucid, Boehringer Ingelheim, GlaxoSmithKline, Janssen-Cilag and MSD. Financial support for research projects was provided by GSK, Johnson & Johnson, Jerini, MSD and Vasopharm. Headache research at the Charité (Berlin) is supported by the German Ministry of Education and Research (BMBF). U. Reuter has no ownership interest in any pharmaceutical company.
Footnotes
Acknowledgements
The authors would like to thank Christine Hofmann and Simon Angele for their help with editing the manuscript.
Appendix
The investigators who recruited subjects for this study are as follows: