
Abstract
Background and objective: Clinical and experimental studies have revealed a central role of calcitonin gene-related peptide (CGRP) in primary headaches. The role of extracellular signal-regulated kinase 1 and 2 (ERK1/2) in neuronal and glial cell expression of CGRP- immunoreactivity (-ir) in rat trigeminal ganglia was studied with an organ culture method.
Experimental procedures: Sections of adult rat trigeminal ganglia were cultured for up to 48 hours, examined with immunohistochemistry and quantitative real-time polymerase chain reaction (PCR) assay. Specific antibodies against CGRP, phosphorylated ERK1/2 (pERK1/2), total ERK1/2 (tERK1/2), phosphorylated p38 (pp38), phosphorylated C-Jun-N-terminal protein kinase (pJNK), pro-calcitonin (pro-CT), CGRP receptor activity modifying protein 1 (RAMP1), glutamine synthetase (GS) and pro-CT were used. To explore molecular mechanisms involved in the organ culture–induced CGRP-ir in neurons and glial cells, the effects of the MEK/ERK1/2 inhibitor U0126, its inactive analogue U0124, the p38 inhibitor SB203580 and the JNK inhibitor SP600125 were studied.
Results: In fresh ganglia, small- and medium-sized neurons were CGRP-ir while some larger neurons displayed RAMP1-ir. Glial cells were negative to both. After organ culture, neurons showed enhanced CGRP- and RAMP1-ir. In addition, some glial cells were RAMP1- and CGRP-ir. Isolated glial cells and neurons were found to contain CGRP mRNA, and showed pro-CT-ir, suggestive of local formation of CGRP. Neurons and glial cells showed enhanced pERK1/2-ir already after two hours of organ culture and this remained elevated for 48 hours. There was transient pJNK-ir in neurons at two hours, while pp38-ir was not altered. U0126 reduced the enhanced pERK1/2-ir, while U0124 had no such effect; the CGRP-ir in neurons and glial cells was reduced at 48 hours and in parallel the CGRP mRNA expression was lower at 24 hours.
Conclusion: We suggest that in conditions of elevated CGRP expression, inhibition of ERK1/2 might be an option for novel treatment.
Introduction
The trigeminovascular system was decoded nearly three decades ago and was shown to harbor several neuronal messenger molecules, such as substance P (SP), neurokinin A (NKA), calcitonin gene-related peptide (CGRP), pituitary adenylate cyclase–activating peptide (PACAP) and the nitric oxide–forming enzyme NO synthase (NOS). It is also recognized that cell bodies in the trigeminal ganglion give rise to sensory C-fibers and mechanosensitive Aδ-fibers that project centrally to the trigeminovascular complex in the brainstem (1).
Currently, migraine is considered to originate in the central nervous system (CNS), and as a result of an acute attack the trigeminovascular system is activated, with release of CGRP at both central and peripheral ramifications with increased levels of CGRP in the cranial venous outflow (2). In support of this view, recent studies have revealed the therapeutic effect of CGRP receptor antagonists (3,4). The site of action of the CGRP receptor antagonists is debated, but presently the peripheral and central ends of the trigeminovascular system are considered to be targets (5), while only limited interest has been directed toward the neurons of the trigeminal ganglion (6). Conditions of intense activation (e.g. application of capsaicin into the mandibular joint) result in strong activation of the mandibular branch (IIIrd) of the trigeminal nerve (7). This is associated with a general intraganglionic spread of activation involving other neurons of the trigeminal ganglion and also the glial cells.
About 40–50% of the trigeminal neurons store CGRP, while neurons containing SP/NKA/NOS/PACAP represent only 10–15% of the total population of neurons (1,8,9). The glial cells constitute more than 90% of the total number of cells in the trigeminal ganglion (7) and they are negative to all the above-mentioned neuronal messengers. Migraine attacks are hypothetically considered to involve a local sterile inflammation in the peripheral ramifications of the trigeminovascular pathway, in the dura mater and the meningeal circulation (10). This hypothesis is supported by observations that inflammatory cytokines may enhance transcription of CGRP in trigeminal neurons (11). Interestingly, triptans have been found to reduce both transcription and release of CGRP from trigeminal neurons (12).
We have recently designed a method to study the putative intraganglionic communications and the interactions between neurons and glial cells using organ culture which induces a local inflammatory reaction with elevation of cytokines (13). In our model, the neurons and glial cells are intact and their connections remain.
The present study was designed to evaluate if organ culture may preserve the tissue elements for up to 48 hours and induce alterations in neuronal messenger and mitogen-activated protein (MAP) kinase expression. We found that the phosphorylated forms of extracellular signal-regulated kinase 1/2 (pERK1/2) MAP kinase were increased in neurons and in glial cells following organ culture. In addition, organ culture induced elevated expression of CGRP in neurons and satellite glial cells (SGCs) via activation of the MAP kinase/ERK kinase (MEK)/ERK1/2 pathway.
Experimental procedures
Organ culture procedure
Adult male Sprague-Dawley rats (weighing 300–400 g) were asphyxiated under CO2 sedation, and the trigeminal ganglia were removed either for organ culture (N = 25) or for control experiments (N = 15). The experiments were approved by the University Animal Ethics Committee (M161-07), Lund University, Sweden.
Trigeminal ganglia were cut rostrocaudally into three pieces (3–4 mm) in ice-cold phosphate-buffered saline (PBS) and transferred to Dulbecco's Modified Eagle's Medium (DMEM) supplemented with penicillin (100 U/mL), streptomycin (100 µl/mL) and amphotericin B (25 µg/mL). The pieces were subsequently incubated for up to 48 hours at 37°C in humidified 5% CO2 in air. In a subset of experiments the trigeminal ganglia were incubated for two hours with exogenous CGRP (10−5 M) (Sigma-Aldrich, Germany) in DMEM and compared to fresh (uncultured) trigeminal sections.
Both incubated and fresh ganglia were immersed overnight in a fixative consisting of 2% paraformaldehyde and 0.2% picric acid in 0.1 mol/L phosphate buffer, pH 7.2. After fixation, the specimens were rinsed in a 10% sucrose-enriched Tyrode solution, snap frozen in cold isopentane and maintained at −80°C.
Intracellular signal-transduction studies
In a subset of experiments, different MAP kinase inhibitors were administered to the culture medium: the MEK1/2 inhibitor U0126 (10−5 M), its inactive analogue U0124 (10−5 M), the p38 inhibitor SB203580 (10−5 M) or the JNK inhibitor SP600125 (10−5 M) (all from Sigma, St. Louis, MO, USA) (N = 12). Control trigeminal ganglia were incubated for the same period and with the same volume (1µL/mL) of the vehicle DMSO (N = 12). Six animals were used for quantitative real-time polymerase chain reaction (qPCR) and five animals from each group were used for immunohistochemistry.
Real-time quantitative PCR
Trigeminal ganglia for real-time PCR experiments were frozen and stored at −80°C until the assays were performed. CGRP mRNA expression levels were quantified after 24 and 48 hours of culture in the presence of the MAP kinase inhibitors U0126 (10−5 M, N = 6), SB203580 (10−5 M, N = 6) and SP600125 (10−5 M, N = 6) or of the vehicle DMSO (N = 6 in each group). The total RNA was extracted and reversely transcribed to cDNA in 40 µl reactions using the GeneAmp reverse transcription kit (Perkin-Elmer, Applied Biosystems, Carlsbad, CA, USA) in a Perkin-Elmer 2400 PCR machine at 42°C for 90 minutes. One µg of RNA was used in each reaction. The real-time qPCR was performed with the GeneAmp SYBR Green PCR kit (Perkin-Elmer, Applied Biosystems) in a GeneAmp 7300 sequence detection system (Perkin-Elmer, Applied Biosystems). The cDNA synthesized described above served as a template. A non-template control was included in all experiments. The PCR reaction was performed in 50 µl and started at a temperature of 50°C for two minutes, 95°C for 10 minutes and the following 40 PCR cycles with 95°C for 15 seconds and 60°C for 1 minutes. Dissociation curves were run after the real-time PCR to identify the specific PCR products. Specific primers for rat CGRP were designed as below:
CGRP forward: 5′-GAGGCAGCTACAAGGTTCAGG -3′
reverse: 5′- AGGTGTTGGTGCTGGACACA-3′
The housekeeping genes glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and elongation factor-1 (EF-1) were used as references in this study due to their continuous expression to a constant amount in cells. Rat GAPDH and EF-1 primers were designed as below:
GAPDH forward: 5′-GGCCTTCCGTGTTCCTACC -3′
reverse: 5′- CGGCATGTCAGATCCACAAC -3′
EF-1 forward: 5′-GCAAGCCCATGTGTGTTGAA-3′
reverse: 5′- TGATGACACCCACAGCAACTG-3′
All primers were designed using the Primer Express 2.0 software (Perkin-Elmer, Applied Biosystems) and synthesized by TAG Copenhagen A/S (Denmark).
Isolation of SGCs for RT-PCR
Fresh or organ cultured trigeminal ganglia were immersed into RNAlater® for 24 hours at 4°C in order to fix mRNA. Thereafter the ganglia were cut into small pieces and transferred to Hanks’ balanced salt solution (pH 7.4), containing collagenase D (0.6 mg/ml; Sigma, St. Louis, MO, USA), papain (1 IU/ml; Sigma) and dispase (2.4 mg/ml; Sigma) (14). After incubation for 40 minutes at 37°C, the tissue were dispersed into a cell suspension by passing through a mesh of 100 µm pore size and gentle centrifuging at 2000 x g for five minutes. The glial-enriched fraction of cell suspension was separated and prepared by a gradient centrifugation (15). Portions (300-400 µl), taken directly from the gradients, were used for RNA extraction and PCR studies as described above.
Immunohistochemistry
Details of primary antibodies used for immunohistochemistry

CGRP = calciton gene-related peptide. MAPK = mitogen-activated protein kinase. RAMP = receptor activity–modifying protein. ERK = extracellular signal-regulated kinase. pERK = phosphorylated ERK. tERK = total ERK. SAPK = stress-activated protein kinase. JNK = C-Jun-N-terminal protein kinase. pJNK = phosphorylated JNK. GS = glutamine synthetase.
Secondary antibodies used for immunohistochemistry

Thereafter, sections were rinsed in PBS-T for 3 x 15 minutes in room temperature and mounted with mounting medium (Crystal Mound, Sigma-Aldrich, Munich, Germany) or Vectashield (Vector Laboratories, Burlingame, CA, USA). Vectashield medium containing 4′, 6-diamidino-2-phenylindole (DAPI, nucleus staining) was used in some sections.
Trigeminal ganglia from additional three rats were placed in 4% paraformaldehyde for two to four hours and subsequently rinsed in raising concentration of sucrose in Sörensen’s phosphate buffer (pH 7.2), embedded in a gelatin medium (30% egg albumin and 3% gelatin in distilled water) and cryosectioned (12 µm). The sections were then treated for immunohistochemistry as described above using a primary antibody against pro-CT (Table 1).
Specimens were examined using an epifluorescence microscope. Co-localization was assessed by super-imposition of separate digital images. Omission of the primary antibody served as negative controls.
Image analysis
Fluorescence was detected at the appropriate wavelength and images of 1392 × 1040 pixels were obtained with 40x objective. The fluorescence intensity was analyzed using the software ImageJ 1.37v (National Institutes of Health, Bethesda, MD, USA, http://rsb.info.nih.gov/ij/). The fluorescence was measured in five areas in each tissue (blinded) using the mean value for each measurements of arbitrary units. The results of controls were set as 100%.
Calculation and statistic
For real-time qPCR experiments data were analyzed with the comparative cycle threshold (CT) method. To evaluate the amount of CGRP mRNA in a sample, GAPDH and EF-1 mRNA were assessed in the same sample simultaneously. The amount of CGRP mRNA was calculated as relative to the amount of GAPDH or EF-1 in the same sample by the formula X0 /R0 = 2CtR-CtX , where X0 = the original amount of CGRP mRNA, R0 = original amount of GAPDH or EF-1 mRNA, CtR the CT value for GAPDH or EF-1 and CtX the CT value for CGRP mRNA.
Statistics were performed using GraphPad 5.0 software (GraphPad, La Jolla, CA, USA) and non-parametric Mann-Whitney U test was applied. p < .05 was regarded as significant. Data are expressed as mean ± standard error of the mean (SEM).
Results
CGRP expression
In fresh (non-cultured) trigeminal ganglia, numerous small- and medium-sized neurons showed cytoplasmic CGRP-immunorectivity (-ir) while the nucleus was spared (Figure 1A–C). CGRP-ir was not found in the glial cells (Figures 1A–C).
Calcitonin gene-related peptide (CGRP)- and glutamine synthetase (GS)- immunoreactivity (-ir) in fresh and incubated trigeminal ganglia. (A) Numerous nerve cell bodies showed CGRP-ir in fresh ganglia (arrows). (B) SGCs, closely surrounding the neurons, were identified with GS (arrows). (C) GS did not co-localize with CGRP in the satellite glial cells (arrows). (D) A large number of neurons (arrowheads) displayed CGRP-ir after 24 hours of culture. SGC nuclei (arrows), and other glial cells nuclei, were seemingly immunopositive for CGRP (arrows), but this was judged to be high-background staining. (E) Arrows point at GS-ir SGCs. (F) Merged image demonstrating GS-ir SGCs (arrows). (G–H) In some areas, intense CGRP-ir SGCs (arrows) were found 24 and 48 hours after culture. SGC = satellite glial cell.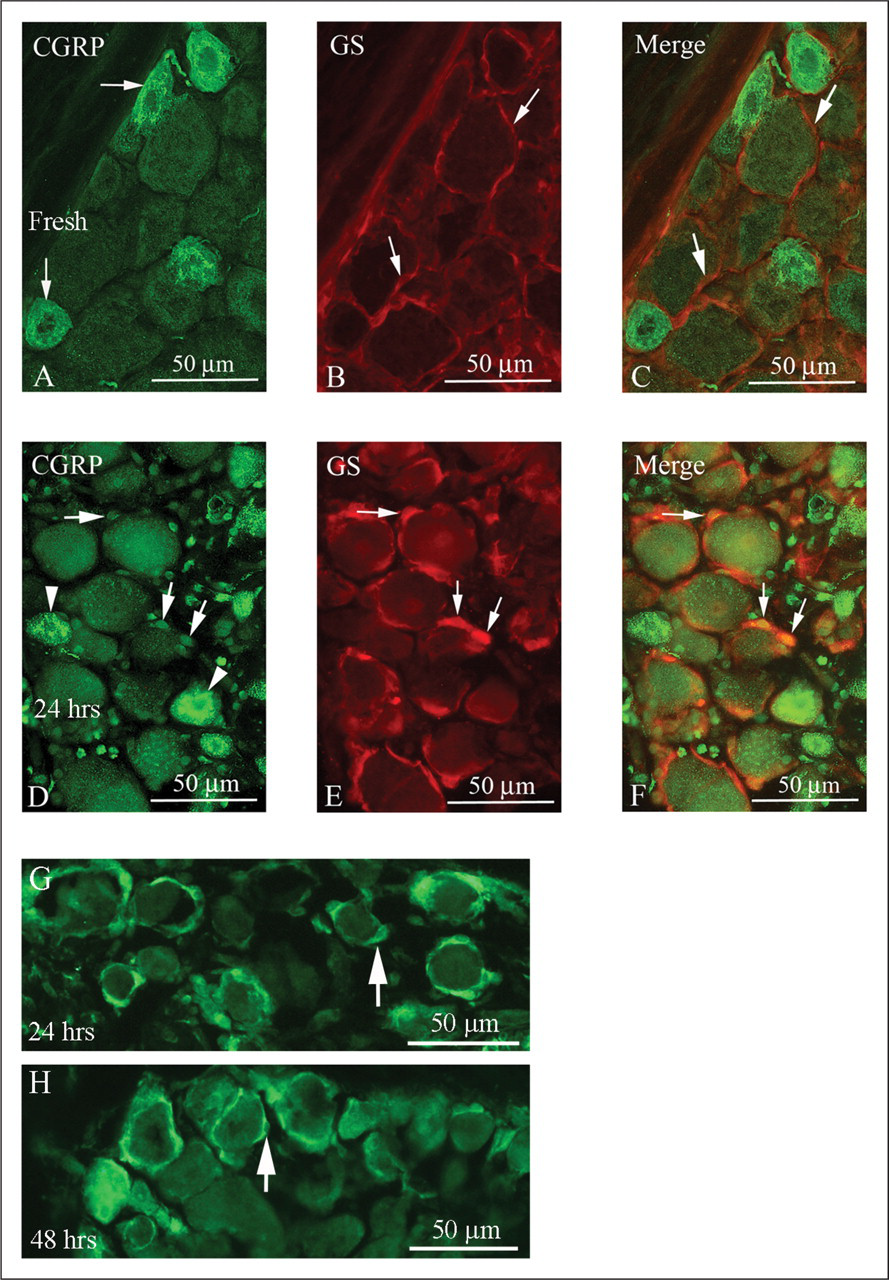
Organ culture for up to 48 hours in serum-free DMEM resulted in enhanced CGRP-ir in the neurons (Figures 1D–F) and in many of the SGCs in some areas of the trigeminal ganglia (Figures 1G–H), often on the surface of the dissected ganglia, indicating association with nourishment. Further culture resulted in deterioration of the tissue and was therefore not studied.
Many SGC nuclei and other glial cells nuclei were seemingly, but weakly, immunopositive for CGRP after culture (Figures 1D,F). However, this finding was judged as un-specific, high-background staining because of the atypical neuronal nuclei staining and morphology that also was found in some areas of the cultured ganglia. In addition, these nuclei were also immunopositive with other primary antisera (e.g. pERK1/2 [see below]).
After organ culture, the CGRP expression in neurons increased by 49% (p < .05) and 72% (p < .01) at 24 and 48 hours, respectively, compared to fresh ganglia (Figure 2).
Comparison of neuronal CGRP-ir in fresh trigeminal ganglion and after organ culture. There was a successive increase in CGRP-ir with culture. Data are given as mean ± standard error of the mean, N = 5. Non-parametric Mann-Whitney U test was applied. *p < .05, **p < .01. CGRP-ir = calcitonin gene-related peptide-immunoreactivity.
In order to evaluate if the increase in CGRP-ir in the SGCs was due to de novo formation, two experiments were performed. First we found that the SGCs have immunoreactivity to pro-CT, a pro-peptide that includes the common region shared by the precursors encoded by the CT and CGRP alternatively spliced mRNAs (16,17). With nuclear DAPI staining we verified that the pro-CT-ir was found in the cytoplasm of the SGCs (Figures 3A–C). In the second experiment, the SGCs were isolated from both fresh ganglia and incubated ganglia after 48 hours of organ culture by enzyme digestion and density centrifugation. The SGCs contained CGRP mRNA which was significantly (p < .01) elevated after organ culture (Figure 3D).
(A–C) Localization of pro-calcitonin (pro-CT) in SGCs in fresh trigeminal ganglion. Co-staining with DAPI (nuclear stain) revealed that the pro-CT-immunoreactivity (-ir) occurs in the cytoplasm of the SGCs (arrows). (D) The CGRP mRNA expression in the SGCs isolated from trigeminal ganglia was significantly increased after 48 hours of organ culture compared to fresh. Data are given as mean ± standard error of the mean, N = 6. Non-parametric Mann-Whitney U test was applied. **p < .01. DAPI = 4', 6-diamidino-2-phenylindole (nuclear staining). SGCs = satellite glial cells. CGRP = calcitonin gene-related peptide.
In order to examine if the enhanced CGRP-ir in the SGCs was due to uptake of CGRP from adjacent neurons, we also performed a series of incubation tests. Co-culture with exogenous CGRP (10−5M) for two hours did not display CGRP-ir in SGCs or show enhanced CGRP expression in neurons.
RAMP1 expression
In fresh trigeminal ganglia, only neurons displayed RAMP1-ir; this was primarily seen in larger neurons, as recently demonstrated (18). Following organ culture, both neurons and SGCs showed expression of RAMP1 (Figures 4A–C). Co-localization experiments with CGRP revealed that there were few neurons positively stained with RAMP1 that also contained CGRP, while neurons positive for RAMP1 most often lacked CGRP.
Demonstration of RAMP1-ir in (A) fresh and (B) cultured rat trigeminal ganglion for 24 hours. Neurons displayed RAMP1-ir (arrows). (C) After organ culture, SGCs exhibited RAMP1-ir (arrowheads). (D) Organ culture for 24 hours displayed enhanced expression of RAMP1-ir in neurons. Data are given as mean ± standard error of the mean, N = 4. Non-parametric Mann-Whitney U test was applied. *p < .05. RAMP1-ir = receptor activity–modifying protein1-(immunoreactivity). SGCs = satellite glial cells.
Intracellular signal studies
In fresh ganglia, neurons and SGCs showed weak immunoreactivity toward the phosphorylated (activated) form of ERK1/2 (Figure 5A), which co-localized with CGRP (Figures 5B–C). Organ culture revealed increased pERK1/2-ir both in neurons and in SGCs. The response was time-dependent with a weak increase already at two hours of organ culture and maintained for the 48 hours period. The pERK1/2 co-localized with CGRP in neurons and SGCs (Figures 5D–F). We observed some pJNK-ir in neurons; however, we did not observe pp38-ir neither in neurons nor in SGCs at two hours. After 24 and 48 hours of culture there was an increase in pJNK-ir, and in addition pp38-ir was found in neurons (data not shown).
Double immunohistochemistry of pERK1/2 and CGRP in fresh trigeminal ganglia (A–C) and after 48 hours of culture (D–F). Co-localization of pERK1/2 and CGRP was found in neurons (arrows) in fresh tissue (A–C). (D–F) Following 48 hours of culture, co-localization was also demonstrated in SGCs (arrows) as well as in the neurons (arrowhead). pERK1/2 = phosphorylated extracellular signal-regulated kinase 1 and 2. CGRP = calcitonin gene-related peptide. SGCs = satellite glial cells.
Incubation of the trigeminal ganglion with the MEK/ERK1/2 pathway inhibitor U0126 (10−5 M) markedly reduced the expression of pERK1/2 (Figure 6) and, in addition, the expression of CGRP in neurons and SGCs (Figures 6D,E). The inactive analogue U0124 (10−5 M) had no inhibitory effect on the elevated CGRP-ir; there were no differences in CGRP-ir, pERK1/2-ir and total ERK1/2-ir by U0124 compared to DMSO (data not shown). The neuronal CGRP-ir was decreased from 100% to 63% at 24 hours (p < .05, Figure 6D) and to 53 % at 48 hours of culture (p < .01, Figure 6D). The CGRP-ir in the SGCs was reduced from 100% to 67% in the presence of U0126 after 48 hours of culture (p < .01, Figure 6E). SP600125 (JNK inhibitor) or SB203580 (p38 inhibitor) treatments did not result in significant changes in the neuronal CGRP-ir.
(A) pERK1/2 staining of fresh trigeminal ganglion. (B) An enhanced expression of pERK1/2-ir was seen in SGCs (arrowheads) and in neurons (arrows) after 24 hours of culture. (C) Co-incubation with the MEK1/2 inhibitor U0126 (10−5 M) markedly reduced the pERK1/2 activity both in SGCs (arrowheads) and in neurons (arrows) at 48 hours of organ culture. (D) U0126 decreased the CGRP-ir expression in neurons after 24 and 48 hours of culture (N = 6), and also in the SGCs (E) at 48 hours (N = 5), but not 24 hours (data not shown). Data are given as mean ± SEM. Non-parametric Mann-Whitney U test was applied. **p < .01. pERK1/2 = phosphorylated extracellular signal-regulated kinase 1 and 2. pERK1/2-ir = pERK-immunoreactivity. SGCs = satellite glial cells. MEK1/2 = MAP kinase/ERK kinase 1/2. CGRP-ir = calcitonin gene-related peptide-immunoreactivity.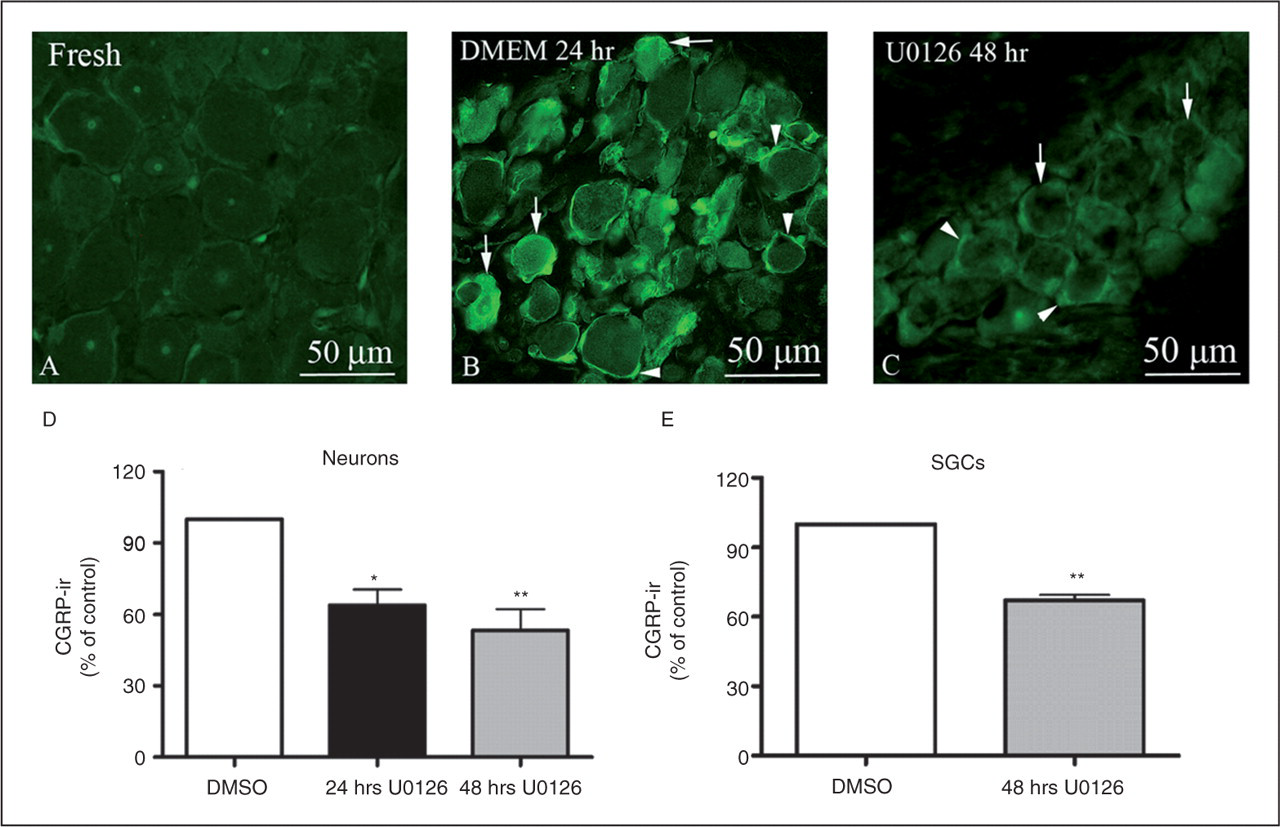
In the presence of U0126 at 24 hours of incubation, the real-time PCR experiments showed reduced CGRP mRNA levels relative to the amount of GAPDH or EF-1 mRNA as compared to control (from 100% to 61% GAPDH, p < .01 or from 100% to 59% EF-1, p < .01) (Figure 7). After 48 hours of culture, U0126 did not affect the CGRP mRNA levels significantly. SP600125 treatment did not result in a significantly lower expression of CGRP mRNA at 24 hours of culture. After 48 hours of culture, the CGRP mRNA levels remained unchanged. Incubation with SB203580 did not show significantly lower expression of CGRP mRNA after 24 and 48 hours of culture (data not shown). Similar patterns of CGRP mRNA expressions were found when using GAPDH as the reference gene, as when using EF-1.
The MEK/ERK1/2 inhibitor U0126 (10−5 M) significantly reduced the CGRP mRNA expression relative to the housekeeping genes glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and elongation factor-1 (EF-1) references after 24 hours of culture of the trigeminal ganglion. Data are given as mean ± SEM, N = 6. Non-parametric Mann-Whitney U test was applied. **p < .01. MEK 1/2 = MAP kinase/ERK kinase 1/2. ERK1/2 = extracellular signal-regulated kinase 1 and 2. CGRP = calcitonin gene-related peptide.
Discussion
The present study has for the first time demonstrated that organ culture, as a model for simultaneous studies of neurons and glial cells in the trigeminal ganglion, results in enhanced expression of CGRP both in neurons and in SGCs. The enhanced expression of CGRP is hypothesized to occur via increased transcription mediated by the MEK/ERK1/2 pathway, already activated at two hours. This view is supported by the observations that fresh glial cells and neurons contain CGRP mRNA; in addition, the SGCs contain cytoplasmal pro-CT. Furthermore, organ culture increased CGRP mRNA and CGRP protein expression.
We here present an organ culture model of trigeminal ganglion, which allows for studies of underlying molecular mechanisms responsible for regulation of neuropeptide expression and interaction in the trigeminal system. Organ culture of trigeminal ganglia sections for up to two days induces over-expression of CGRP in neurons. We must first and last clarify that this is not a physiologic method, because the organ culture is a way to induce stress to the system which is accompanied by a rise in local cytokines (13); it is a way to activate inflammatory mechanisms within the trigeminal ganglion. Thus, the intracellular signal studies revealed that the enhanced expression of CGRP-ir occurs via early phosphorylation of ERK1/2. The specific MEK1/2 inhibitor (U0126), but not its inactive analogue U0124, significantly reduced the enhanced pERK1/2 expression and the CGRP-ir in neurons and in SGCs at 48 hours in parallel with an early reduced CGRP mRNA expression at 24 hours. Furthermore, total ERK1/2 was unchanged. This demonstrates a time sequence of intracellular events; organ culture of trigeminal ganglion induces early activation of ERK1/2 and a subsequent enhanced expression of CGRP mRNA, which is followed by protein expression in the cells. Other MAPK pathways were not very active; at this early time point there was only a transient pJNK and no change of pp38 visible.
MAP kinases consist of three main pathways (ERK1/2, p38 and JNK) (19), which can be activated by various stimuli such as stress, cytokines and growth factors (20–22). The proinflammatory cytokines tumor necrosis factor-α (TNF-α) and interleukins (ILs) can activate MAP kinases (23) and increase CGRP transcription (11,24). Under conditions mimicking neurogenic inflammation in vitro, the CGRP transcription is increased (11,25).
During a migraine attack the trigeminal nerves release CGRP (2), which in turn may cause the production and release of endogenous pro-inflammatory mediators (26). The presently observed increase of CGRP in neurons and SGCs is suggested to be mediated by MAP kinases (12,27). The phosphorylation of ERK1/2 in the dorsal root ganglion and in dorsal horn neurons is increased in response to noxious stimulation of peripheral tissue (28,29) or upon electrical stimulation to the peripheral nerve (30). The anti-migraine drug sumatriptan can block MAP kinase activation via sustained elevation of intracellular calcium in cultured trigeminal neurons (12). In concert with this, we observed that the MEK1/2 inhibitor U0126 significantly attenuated the increase in ERK1/2 activity, and suppressed the expression of CGRP mRNA and CGRP-ir both in neurons and in glial cells. Recently it was observed in vivo that capsaicin administration increased the pERK1/2 activity in the trigeminal ganglion and that capsaicin significantly increased the expression of the c-fos in the spinal trigeminal nucleus (31). The pre-treatment with a CGRP receptor antagonist prevented the capsaicin-induced activation, which lends further support for the role of the trigeminal system in primary headaches.
A recent study has reported on the existence of neuronal–glial communications via gap junctions and paracrine signaling, suggesting a role in the development of sensitization within the trigeminal ganglion (7). However, in the present set-up, the increased CGRP-ir in the glial cells did not appear to be due to a transitory release and uptake from neurons via gap junctions into glial cells, as the glial cells did not show an increase in CGRP-ir after incubation with exogenous CGRP and thereby functionally link glial cells and neurons. A direct diffusion from neurons to glial cells is also unlikely, provided that CGRP is stored in vesicles. In addition, SCGs, which exhibit CGRP-ir, were seen around both neurons that were positive for CGRP-ir and neurons that were negative for CGRP, suggesting that their CGRP-ir did not emanate from the trigeminal neurons. This view is strongly supported by the real-time PCR studies, which show that the glial cells express CGRP mRNA in both fresh and cultured SGCs. The occurrence of mRNA and not the protein is common in biology. In addition, the SGCs were positive for pro-CT-ir, the precursor of CGRP. It was early demonstrated that the calcitonin gene produces CT in the thyroid but CGRP in neuronal tissue (16,17). In agreement with the CGRP protein studies, the expression of CGRP mRNA by the glial cells is increased significantly after organ culture.
In addition, we observed enhanced RAMP1-ir in neurons and glial cells after culture. The localization of the CGRP receptor element RAMP1 in CGRP-containing or -lacking neurons, furthermore, suggests that neuronal or intraganglionic release of CGRP may act within the trigeminal ganglion on receptors with different locations and may activate the CGRP-negative neurons in the trigeminal ganglion. Recently it was shown that trigeminal neurons in humans and rats co-localize calcitonin-like receptor (CLR) and RAMP1, the two components that form the mature CGRP receptor (18). The interaction between CGRP and CGRP receptors in the trigeminal system may play a role in the development and perpetuation of primary headaches but could equally well be involved in other cranial inflammatory and neuropathic conditions.
In conclusion, the present findings indicate that activation of a MAP kinase–dependent inflammatory signal pathway is involved in over-expression of CGRP in nociceptive neurons and could participate in generating pain hypersensitivity. We have for the first time demonstrated that organ culture of the trigeminal ganglion induces activation of ERK1/2, with an enhanced expression of CGRP in both neurons and glial cells.
Footnotes
Acknowledgements
We are grateful for the suggestion of Professor Andrew Russo to study pro-calcitonin and for the help with these experiments provided by Professor Karin Warfvinge and Warfvinge Science Support AB. This study is supported in part by grants from the Swedish Research Council (no. 5958), the Lundbeck Foundation and the Hungarian Ministry of Health (ETT: 210/2006).
Disclosure
JT, AK and LE carried out the experiments, participated in design, statistical analysis and writing of the manuscript. LE and CBX conceived the study and participated in its design, coordination of experiments and the writing. All authors participated in the writing of the manuscript and approved the final manuscript.