
Abstract
Kikuchi-Fujimoto disease is a rare, benign cause of necrotising lymphadenitis often presenting with fever. We describe a case of a 17-year-old boy with non-verbal autism presenting to our intensive care unit with prolonged fever of unknown cause. This case highlights the role of the intensive care unit in cases of diagnostic dilemma. The critical care community should be aware of Kikuchi-Fujimoto disease as although it is usually benign, it can rarely lead to acute airway compromise.
Keywords
Introduction
Kikuchi-Fujimoto disease (KFD) is an uncommon disease of unknown aetiology characterised by cervical lymphadenopathy, fever, rash, arthritis and fatigue. The aetiology of KFD is unknown but it can be associated with autoimmune disease including systemic lupus erythematosus (SLE) and bacterial, viral, fungal and parasitic infectious diseases.
Case report
A 17-year-old boy presented with a three-week history of intermittent fevers up to 39°C, cough and watery stools. He had a background of non-verbal autism, was born in Bangladesh and immigrated to Australia at the age of two. His only regular medication was atomoxetine 60 mg daily but he had recently been prescribed amoxicillin for a presumed respiratory infection. On admission he was lethargic, agitated and febrile. Clinical examination was limited due to his behavioural disorder. He was treated empirically with ceftriaxone, which was broadened to piperacillin-tazobactam and azithromycin for presumed respiratory sepsis but he continued to deteriorate. Due to persistent pyrexia of greater than 39°C with associated hypotension and tachycardia he was transferred to the intensive care unit (ICU) for closer monitoring, cooling and consideration for vasopressor support.
Investigations
The most significant laboratory findings included neutropenia, progressive derangement of his liver function and increasing levels of ferritin and lactate dehydrogenase (LDH) (Figures 1 and 2). He was comprehensively investigated for potential infectious causes as detailed in Table 1 but the diagnosis remained elusive. Of note he was weakly positive for parvovirus B19 IgM (sample to cut-off ratio of 1.2), with a negative IgG. An autoimmune screen was entirely unremarkable. Computed tomography (CT) imaging of his brain, chest and abdomen was performed and demonstrated minimal mediastinal and hilar reactive lymphadenopathy only. There was no evidence of endocarditis on a trans-oesophageal echocardiogram. Lumbar puncture demonstrated normal cerebrospinal fluid and negative results to all pathogens.
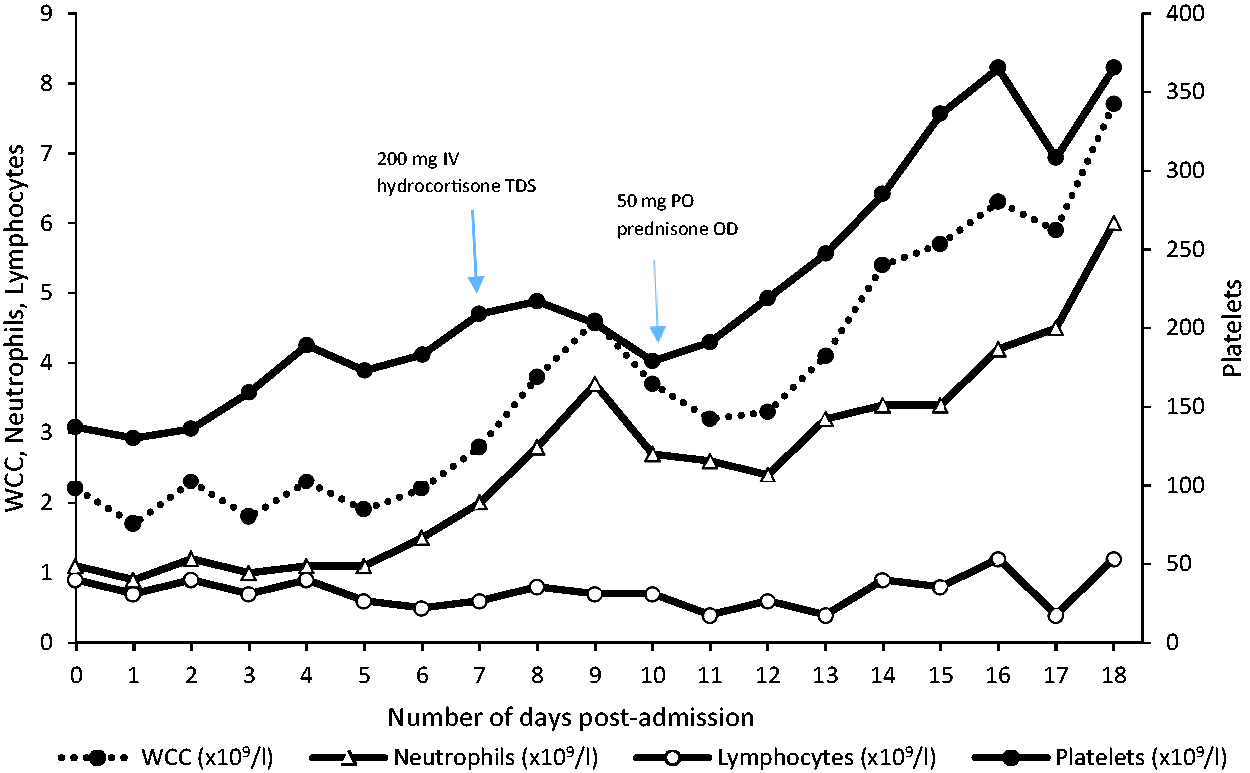
Trend for common markers of Kikuchi-Fujimoto disease in our patient. WCC: white cell count; IV: intravenous; TDS: ter die sumendum (three times per day); PO: per os; OD: once daily.
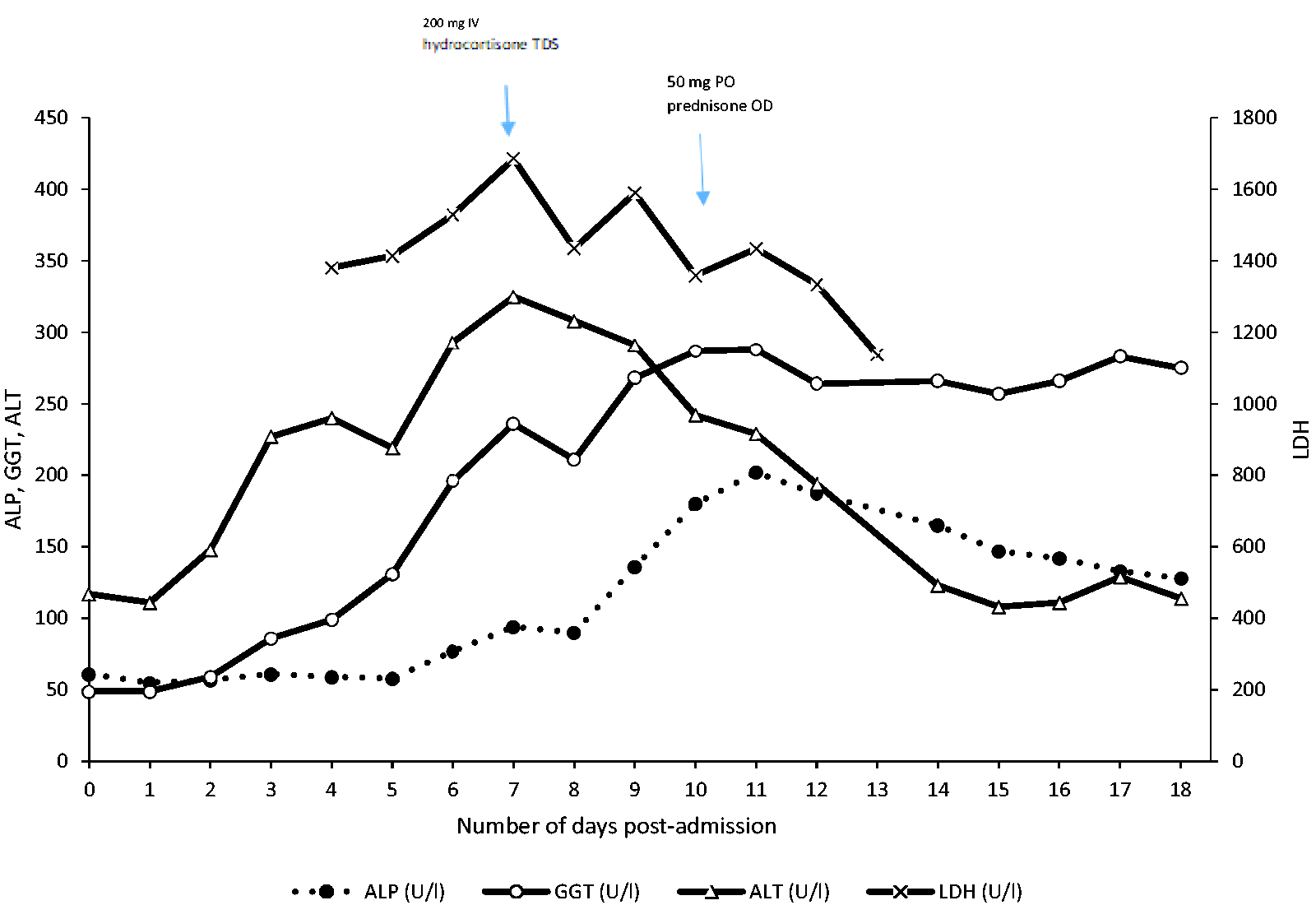
Trend for non-specific markers of Kikuchi-Fujimoto disease in our patient. ALP: alkaline phosphatase; GGT: gamma-glutamyl transferase; ALT: alanine aminotransferase; LDH: lactate dehydrogenase.
Results of infectious screen.

HIV: human immunodeficiency viruses: IgG: immunoglobulin G; IgM: immunoglobulin M; CMV: cytomegalovirus; EBV: Epstein-Barr virus; IGRA: Interferon Gamma Release Assay; CSF: cerebrospinal fluid; TB: tuberculosis; DNA: deoxyribonucleic acid.
Progress
The patient developed an acute exanthematous maculopapular rash; skin biopsy revealed a non-specific interface dermatitis, with an occasional Civatte body (necrotic keratinocytes) with a mild lymphocytic infiltrate. The direct immunofluorescence was negative with no evidence of skin involvement secondary to SLE.
Considering the above, the diagnosis of haemophagocytic lymphohistiocytosis (HLH) and a primary haematological malignancy was considered. However, his bone marrow biopsy demonstrated normocellular marrow with mild to moderate haemophagocytosis; these changes were felt to represent a reactive process.
On day 6 of ICU admission, we noted a rapidly expanding unilateral cervical swelling and an urgent repeat CT of the neck revealed new widespread lymphadenopathy with a heterogeneous appearance in keeping with necrosis (Figures 3 and 4). A clinical diagnosis of KFD was suspected and a core biopsy of cervical lymph nodes was performed (see Figure 5). The histopathological examination of the lymph nodes was consistent with KFD and the patient was commenced on 200 mg hydrocortisone three times per day with a rapid resolution of symptoms. He was successfully weaned from ventilation and extubated eleven days after admission to ICU. After a period of rehabilitation, he was discharged on day 34 of admission with a steroid tapering regimen.
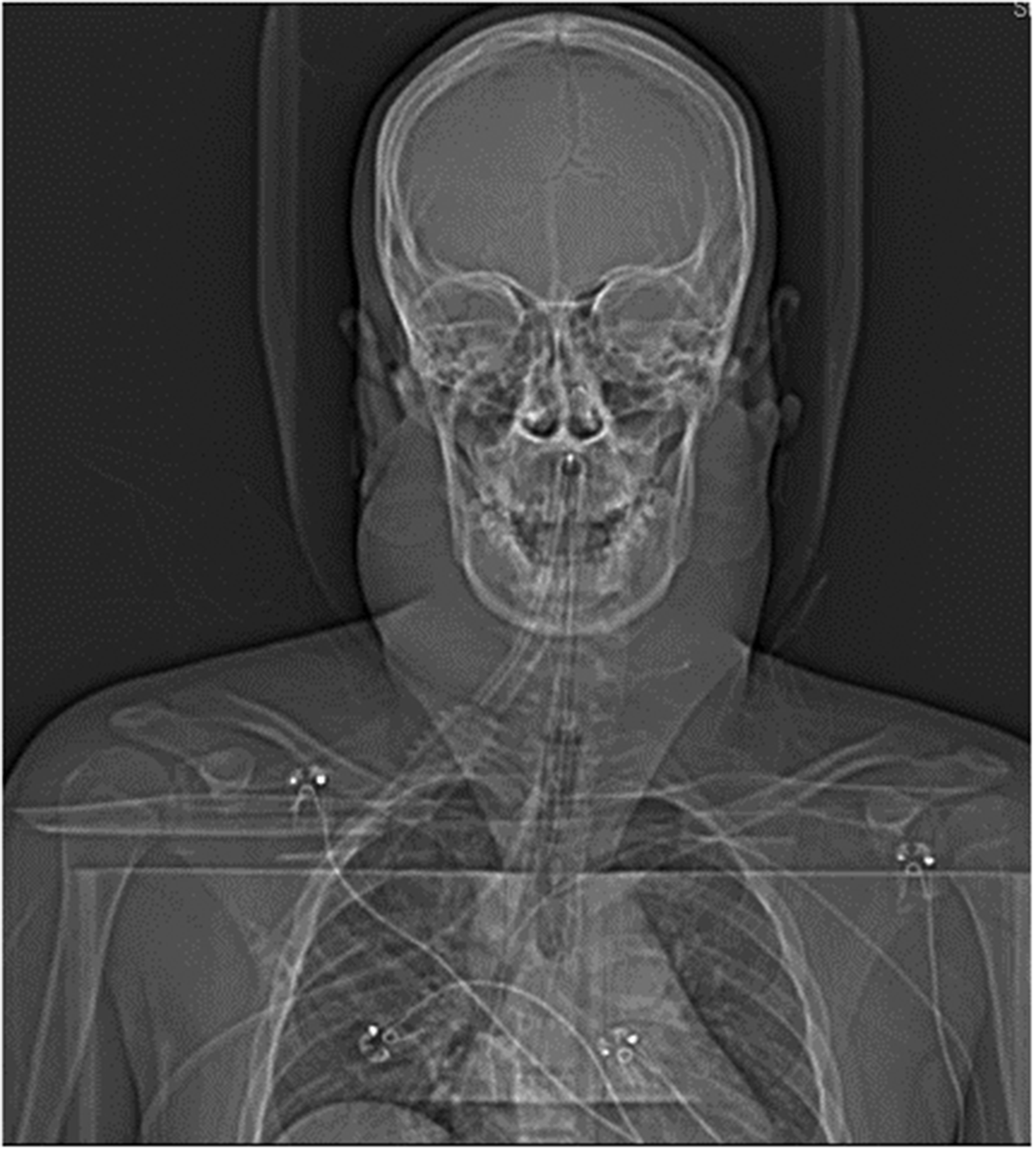
Coronal computed tomography demonstrating widespread lymphadenopathy.
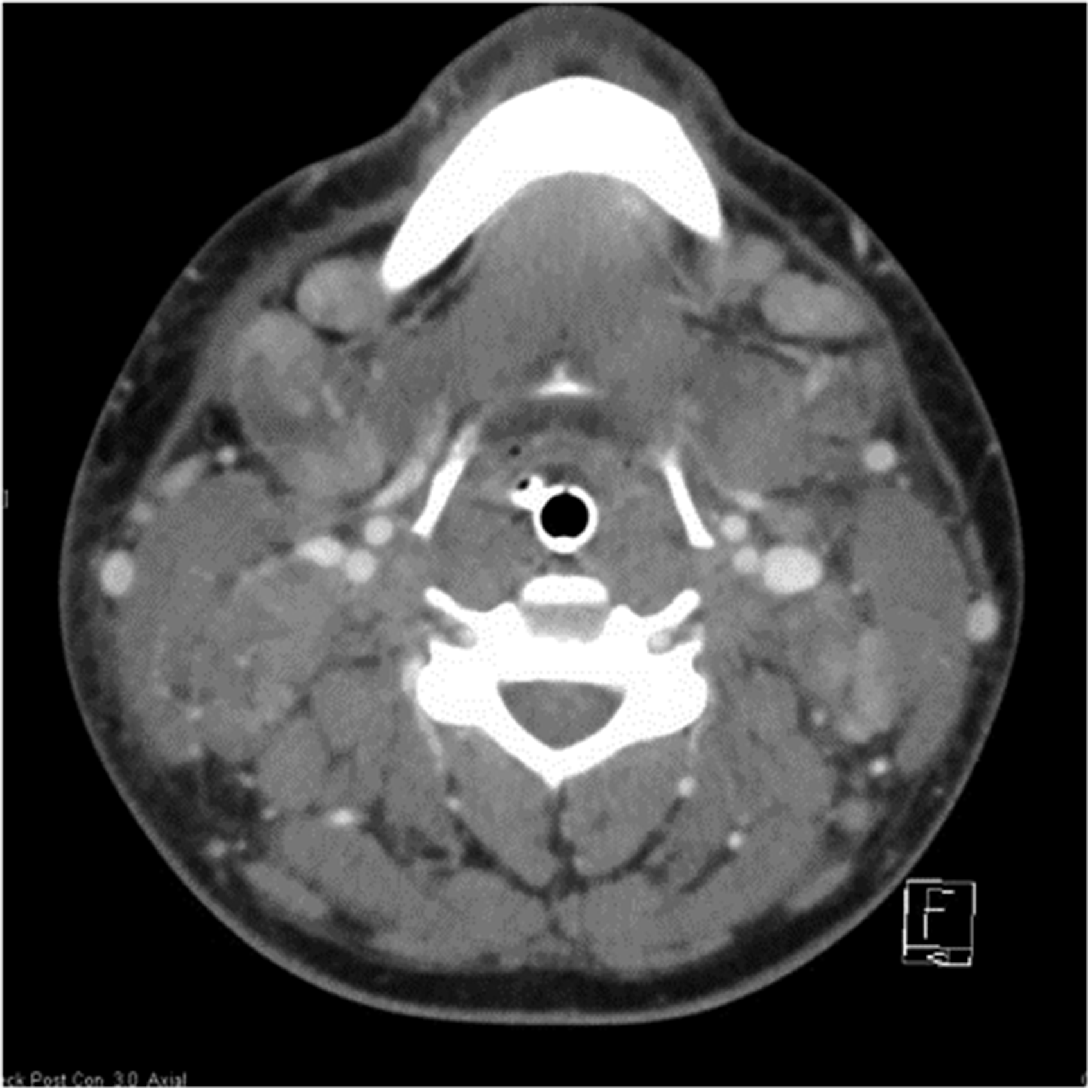
Transverse computed tomography demonstrating multiple small and medium-sized lymph nodes.
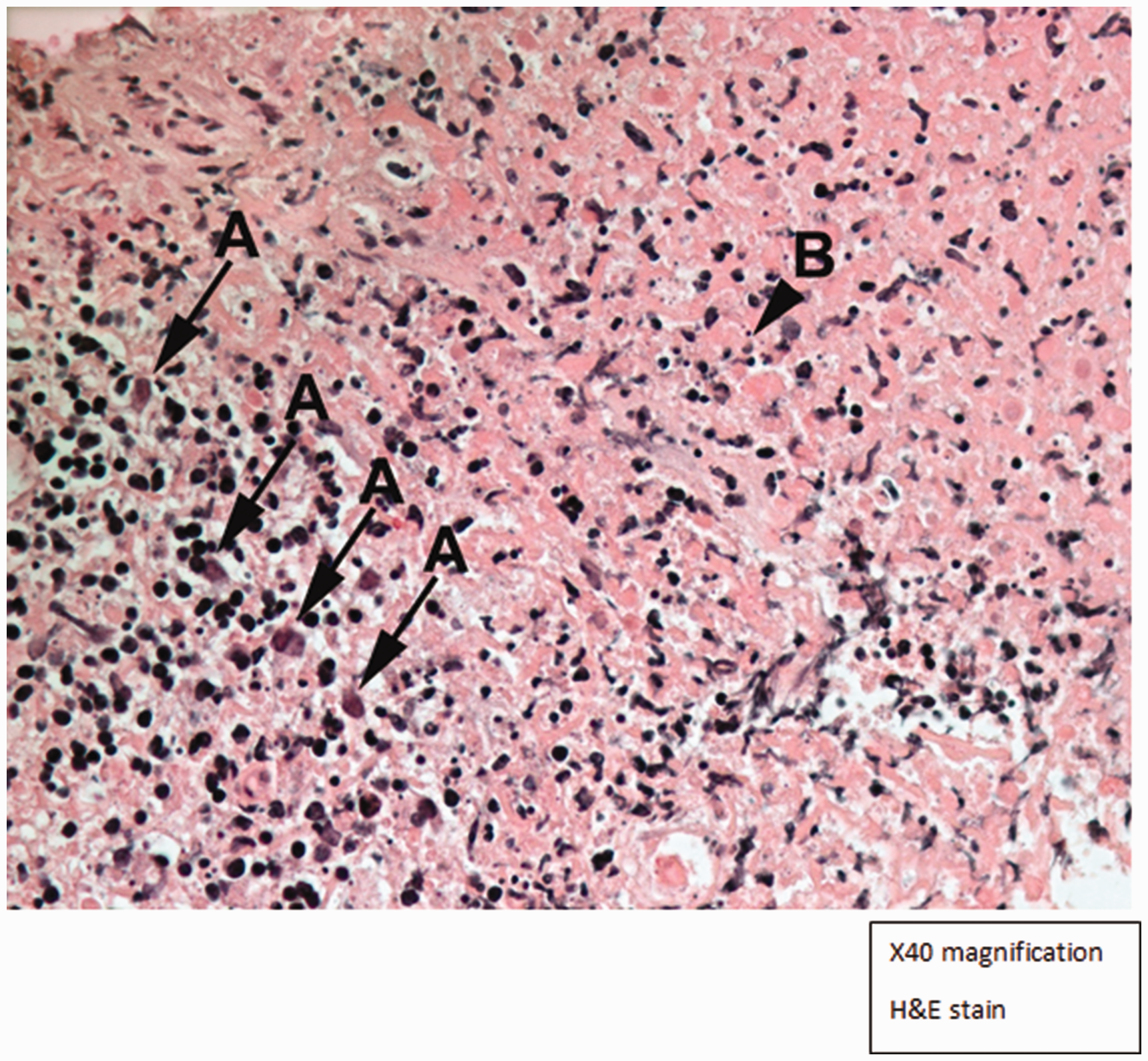
Histological image of cervical lymph node biopsy demonstrating characteristic features of Kikuchi-Fujimoto disease. (a) Plasmacytoid dendritic cells; (b) widespread necrosis with karyorrhexis and absent neutrophils.
Unfortunately, he re-presented three weeks after discharge while on 30 mg of prednisone daily. Re-escalating the prednisolone dose back to 50 mg daily with the addition of hydroxychloroquine enabled rapid remission. He was subsequently successfully weaned off both prednisone and hydroxychloroquine over six months and remained well eight months later with no further relapses.
Discussion
KFD was first described in 1972 by Dr Kikuchi and Dr Fujimoto with two cases of lymphadenitis with histological features of paracortical necrosis and mononuclear cell reaction.1,2 Previously thought to be more common in young Asian women,3–5 it has now been reported worldwide in both men and women.6–8 KFD may present acutely or subacutely and is characterised by lymphadenopathy, fever and respiratory symptoms. The most common clinical sign is cervical lymphadenopathy, which occurs in up to 80% of cases.9–12 Although rare, if pronounced lymphadenopathy occurs acutely, patients may present with features of superior vena cava obstruction or airway obstruction. 13
Reaching a diagnosis of KFD can be challenging as there are no defined serological criteria and blood tests can be normal. The most common abnormal findings are leukopenia, neutropenia, lymphocytosis or thrombocytopenia. 14 Other non-specific findings demonstrated here include transaminitis, elevated LDH and moderately elevated ferritin. 15 However, unusually this patient also had elevated triglycerides, not typically associated with KFD but usually with either adult onset Still's Disease or HLH. Lymph node biopsy therefore is gold standard and diagnosis is confirmed with a triad of characteristic histological changes that were seen here. These are (a) necrotising lymphadenitis; (b) widespread karyorrhexis within the necrotic areas; and (c) predominance of mononuclear cell infiltrate with occasional macrophages and absent neutrophils.9,10,12
Although the pathogenesis is not clear, KFD can be associated with infectious or autoimmune aetiology. Infectious causes may include Brucella, Bartonella henselae, Yersinia enterocolitica, Toxoplasma gondii, fungal disease such as histoplasmosis, herpes viruses, human immunodeficiency viruses, Epstein-Barr virus and rubella. 16 There are reports in the literature of parvovirus B19-associated KFD; in this case, the significance is unclear as he was initially weakly positive for parvovirus B19 IgM but on repeat testing three weeks later he remained immunoglobulin G (IgG) negative and IgM positive. This is suggestive of a false-positive IgM result. There is also strong link between KFD and underlying autoimmune disease, particularly SLE; KFD can precede or overlap with the diagnosis of SLE and screening for autoimmune disease should be considered in any patient with KFD. 17
There are no unifying treatment guidelines, but supportive treatment (primarily non-steroidal anti-inflammatories and antipyretic agents) and observation is often sufficient for mild cases.16,18 If a patient requires treatment the mainstay is either steroid therapy or steroid-sparing agents including hydroxychloroquine. 18 Case series report either high-dose oral prednisone at 0.5–1 mg/kg or high-dose methylprednisolone. Intravenous immunoglobulin has been successfully used in patients with severe disease with good results. 19 Overall the outcome of KFD is usually favourable. KFD patients can relapse and although it is difficult to assess the true rates of recurrence, reports vary from 3%–5%20,21 and can be underestimated due to inadequate long-term clinical follow-up. Dumas et al. 18 reported that of 91 patients, 61.5% improved without treatment. As reported by Deaver et al., 16 mortality rates are low between 0.5% and 2.1% and many cases run a relatively benign course. To our knowledge, this is one of very few case reports of a patient with KFD requiring ICU admission.
Conclusion
KFD needs to be considered in the differential diagnosis in patients presenting with prolonged fever of uncertain aetiology particularly in the context of marked lymphadenopathy. KFD may present either as part of the clinical spectrum of specific autoimmune diseases, most commonly SLE, or as an enigmatic idiopathic condition as illustrated by this case report. The management should involve a multidisciplinary team including experienced histopathologists. Prompt recognition of this condition may avoid unnecessary treatments for patients and enable rapid resolution.
Footnotes
Author contributions
Helena Crawshaw: conceptualisation, formal analysis, writing of original draft, review and editing. Kanaka Sundaram Rachakonda: conceptualisation, supervision, review and editing. Leah Kim: conceptualisation, project administration, writing of the original draft. Thulasi Jegatheesan: project administration, supervision and validation. Alar Enno: formal analysis, investigation, visualisation, review and editing.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
A statement of consent was gathered from the patient’s legal guardian (his mother) to publish this case and the associated images.