
Abstract
Through an analysis of the FDA’s approval of the controversial anti-influenza drug Relenza (zanamivir), we interrogate distinct social scientific theories of pharmaceutical regulation. We investigate why, despite internal negative opinions and an Advisory Committee’s non-approval recommendation, the FDA approved Relenza in the late 1990s. Based on a close reading of FDA documents, we show how agency officials guided the manufacturer’s analyses and participated in constructing a tenuous argument for approval. We show how regulators may strategically design drug labels that can justify their permissive regulation. We consider the explanatory power of official accounts and alternative, partially overlapping, theories of pharmaceutical regulation in the Relenza case, and develop new insights into the institutional dynamics of regulator-industry relations. We find little or no evidence that the FDA was primarily driven by public health concerns, pressure from disease-based patient activism, or a consumerist and neoliberal regulatory logic, although some of these explanations provided managers with convenient rhetoric to rationalize their actions. Rather, we argue that the Relenza case highlights contradictions between a scientific culture at FDA, conducive to rigorous product evaluations, and the agency’s attempts to accommodate higher-level political (i.e. Congress) and industry demands conducive of permissive regulation – consistent with some aspects of reputational and capture theories, as well as with corporate bias theory.
Keywords
Introduction
A major task of the United States Food and Drug Administration (FDA) is to review the scientific evidence supporting new drug applications (NDAs) submitted by companies applying for market approval. If the FDA decides that marketing approval should be granted, it must also decide – in dialogue with the manufacturer – how to describe the drug’s pharmacological, toxicological and clinical properties to health professionals. This FDA approved information is contained in a document known as the product label, whose stated purpose is to convey clear, informative and accurate drug information with relevance to US doctors and patients (FDA, 2006).
The FDA’s ‘gatekeeping’ power – its decisions to grant or withhold marketing approval for drugs – has enormous implications for public health and the broader healthcare system. Equally, the FDA’s oversight of the content and claims made in drug labels are important for public health and the broader healthcare system, since labels contain critical prescribing information for clinicians and determine the legal boundaries for companies’ marketing claims (Griffin et al., 2012). However, the label may also be used strategically to protect permissive, rather than precautionary, regulation, where regulators focus on communicating risks or uncertainty, rather than rejecting or withdrawing drugs on safety or efficacy grounds (Davis and Abraham, 2013). Additionally, because companies are involved in drafting labels, and because they have an apparent interest in labeling that would authorize broad marketing claims, they are likely to suggest language that allows for more, rather than less, optimistic interpretations of their drugs’ therapeutic value. Taken together, investigating how regulators and companies negotiate and use labeling is likely to offer fresh insights into how social factors intersect with technical data on drugs to shape regulatory assessment and outcomes – and hence may advance our understanding of pharmaceutical regulation.
Here we utilize process-tracing (Bennett and Checkel, 2015) to investigate FDA’s controversial approval and labeling of the first neuraminidase inhibitor-type anti-influenza drug, Relenza, in 1999. We aim to account for the FDA’s approval and labeling of Relenza, first, through an in-depth exploration of the regulatory process that preceded approval and, second, by considering the explanatory power of both official accounts and alternative social scientific theories of pharmaceutical regulation in the Relenza case. Finally, we consider the implications of this case study for the current regulatory environment in the US.
Research problem and theoretical background
In July 1999, the FDA approved Relenza (zanamivir) for the treatment of influenza, and in the wake of regulatory approval of Relenza and its better known cousin Tamiflu (oseltamivir), governments across the globe spent huge amounts of money stockpiling these drugs. Yet in 2014 the Cochrane collaboration published meta-analyses showing that the drugs’ efficacy had been overstated, in part because of major reporting and publication biases (Jefferson et al., 2014). This resulted in accusations that the manufacturers and manufacturer-funded academics had exaggerated treatment benefits for commercial gain, and criticism of governments for having allowed themselves to be misled into stockpiling drugs with limited efficacy (Jefferson and Doshi, 2014).
Questions were also raised about the FDA’s initial approval of Relenza, with one prominent medical journalist describing it as ‘a case study in failed regulation’ (Moynihan, 2014: 1). However, in another article we show how the FDA had in fact documented many of the weaknesses that characterized the evidence base for Relenza in the late 1990s, and explain this in terms of a distinct scientific culture that encouraged FDA reviewers to adopt a meticulous ‘bottom up’ approach to scrutinizing company applications – an approach that often involved independent examination of trial documentation and statistical reanalysis of raw data (Mulinari and Davis, 2017). A key puzzle, then, is why the FDA approved Relenza in the face of FDA scientists’ doubts over whether trials demonstrated that the drug offered meaningful benefits to patients.
To address this puzzle, we investigate the FDA’s approval of Relenza and consider the explanatory power of key frameworks drawn on by social scientists and other commentators to explain trends and outcomes in pharmaceutical regulation. These are: public interest theories, capture and corporate bias theories, disease-politics and reputational theories, and a distinctly consumerist and neoliberal regulatory logic. These frameworks are not mutually exclusive, nor do they necessarily represent coherent traditions or approaches. Nevertheless, scholars with a disease-politics perspective, for example, point to the same key factor (patient activism) to explain regulatory outcomes and apparent shifts in the FDA’s philosophy and practice since the late 1980s (Daemmrich, 2004; Edgar and Rothman, 1990). And scholars drawing on capture (Carpenter and Moss, 2014) and corporate bias (Davis and Abraham, 2013) theories emphasize industry influence and power as dominant factors shaping organizational behavior and outcomes. In the sections below, we outline some of the key features of these frameworks and how they might be pertinent to FDA’s review of the Relenza NDA.
Public interest theories of regulation
Public interest theories of regulation (Posner, 1974) – which have their roots in the progressive law and economics scholarship of the late nineteenth century (Fried, 2001; Novak, 2017) – underpin ‘common-sense’ and official accounts of regulation, with some broad notion of the public interest often explicitly articulated in the mission statement of regulatory agencies. During its review of the Relenza application, the FDA offered a number of justifications for its regulatory actions, which we discuss in detail below. A key justification rested on the FDA’s claim that there was a pressing public health need for anti-influenza drugs. Echoing this claim, Elbe (2018) has suggested that the FDA approved Relenza because the perceived health and security threats of influenza pandemics trumped agency concerns about limited drug efficacy. Such an explanatory framework would include the possibility that the FDA reached the ‘wrong’ decision in approving Relenza, but this would be because the FDA misjudged where the public health interest lay in this particular case.
Capture and corporate bias theories
A diametrically opposed perspective, suggested by a spate of commentaries on the FDA that appeared in the medical and the lay press around the time of Relenza’s approval, is that the FDA is increasingly likely to act in the interests of the pharmaceutical industry, rather than in the interests of public health – in part as a consequence of the introduction in the US in 1992 of ‘user fees’ and the agency’s increasing dependence on industry funding (e.g. Frontline, 2003; Horton, 2001; Willman, 2000). Indeed, this was the interpretation suggested by Dr Michael Elashoff, the FDA statistical reviewer who worked on the Relenza NDA. In a 2003 interview with Public Broadcasting Service’s investigative journalism program, Frontline, Elashoff states: When I started in 1995, I didn’t perceive the same level of pressure [to approve new drugs] as I did when I left in 2000. So definitely something changed during the five years I was there, and that also coincided with the User Fee Act. (Frontline, 2003)
Accusations of inappropriate industry influence over regulatory agencies are often framed in terms of the academic concepts of regulatory capture or corporate bias. In contrast to public interest theories of regulation, both capture (Carpenter and Moss, 2014) and corporate bias theories (Davis and Abraham, 2013) suggest that regulators will systematically prioritize the interests of the industry. However, corporate bias theory is distinct from traditional capture theory to the extent it seeks to situate regulatory decision-making within the broader political context of state-society relations, looking beyond the immediate relationship between regulator and regulated industry. Specifically, corporate bias theory postulates that certain organized interests (for instance, the pharmaceutical industry) are able to gain privileged access to the state, such that ‘those organized interests have governed in partnership with the state, setting the agenda for regulation’ in a way that systematically biases regulation ‘in favour of those interests [and] at the expense of diverging or conflicting interests’ (Davis and Abraham, 2013: 33).
There is a growing body of work within the social studies of pharmaceuticals regulation in Europe, North America and Australia that, in linking the macro- and meso-level politics of regulatory change to the micro-sociology of regulatory decision-making, has provided support for corporate bias theory (Abraham, 1995; Davis and Abraham, 2013; Lexchin, 2016; Löfgren and de Boer, 2004; Ozieranski and King, 2017). Similarly, in his analysis of the history of US medicines regulation, Carpenter (2010: 731) argues that by the late 1990s FDA behavior was changing following an ‘organized conservative assault’ on the agency from within and outside of Congress and suggests that this may have weakened regulatory independence and the ability to uphold traditional standards of efficacy and safety. However, it does not follow from this that corporate bias or capture theories are able to account for every instance of regulatory decision-making. And indeed, there are other dominant (and sometimes competing) frameworks that have sought to explain trends and outcomes in pharmaceutical regulation, and these must also be considered.
Disease-politics and reputational theories
Disease-politics theories postulate that, since the AIDS crisis, it is primarily the demands and activism of disease-based patient groups that have altered drug regulation in the US (Daemmrich, 2004; Edgar and Rothman, 1990; Vogel, 2012). Specifically, disease-politics theories contend that patient activism has pushed the FDA to be less ‘risk-averse’ and to accept greater uncertainty with respect to drugs’ efficacy and safety. While disease-politics theories may be agnostic over whether these regulatory shifts were in the interests of public health (Edgar and Rothman, 1990), some senior FDA officials have sought to explain changing regulatory philosophy and practice by integrating public interest claims with a disease-politics perspective. For example, one FDA manager stated: There are some situations where you promote public health by protecting people and other situations where you promote health by being a facilitator and making sure they get what they need, and making sure you’re not being a roadblock . . . And it was that piece of the puzzle that the Agency had been blind to . . . And that’s really what the HIV population woke this agency up to, philosophically. (in Davis and Abraham, 2013: 37)
Reputation-based theories share some common ground with disease-politics theories in that Carpenter (2004) has argued that, since the 1980s, ‘patients, more than pharmaceutical firms, shape the political costs to the FDA of delaying drug approval’ (p. 52). According to Carpenter (2004: 61), ‘The FDA protects its reputation . . . and responds dramatically to patient advocacy groups and their coverage in the media because they make the consequences of delay and rejection more visible.’ However, Carpenter’s (2010) elaboration of reputation-based regulation goes further in theorizing a ‘politics of reputation’, in which regulatory power derives primarily from agency reputation (Carpenter, 2010: 730), so that managing, maintaining and enhancing FDA’s public image – through its responses to and interactions with various ‘audiences’ across science, government, industry, health care and disease-based patient advocacy groups – becomes a paramount agency goal. In this way, a reputational approach may also be consistent with capture or corporate bias theories of regulation – since regulatory acquiescence vis-à-vis industry may represent self-preserving attempts by FDA to fend off attacks that threaten its reputation (Hilts, 2003). However, reputation-based accounts have tended to be offered as alternative explanations to claims that FDA is dominated or captured by industry interests (Carpenter, 2004, 2010).
Neoliberal regulatory logic and consumer choice
A further perspective within the social science literature suggests that the more individualistic, anti-government and consumerist North American culture might mean that US regulators are likely to adopt a more consumer-oriented approach to their gatekeeping role than, for example, their European counterparts (Bodansky, 2003; Levy and Newell, 2000). On such a view, where evidence in support of the safety and efficacy of a new drug was borderline or equivocal, as happened in the Relenza case, the FDA would be more likely to approve that drug, ensuring however that the product label provided sufficient information for clinicians and patients to make informed decisions about use (Lievre, 2002).
If it is true that US culture is distinctly consumerist, then for much of its history the FDA existed in a paradoxical relationship with that culture, since it was widely acknowledged to offer greater protection to the public, or (according to its critics) to be more paternalistic, than other national drug regulatory agencies (Abraham, 1995; Carpenter, 2010; Daemmrich, 2004; Edgar and Rothman, 1990; Hilts, 2003). However, Vogel (2012) notes that from 1990 onwards the trajectory for US regulatory policies with respect to consumer safety and the environment was to offer less protection to the public than in Europe. In the 1990s, this shift towards more permissive regulation occurred against a political backdrop in the US in which the Republican party had adopted an increasingly anti-government and deregulatory rhetoric (Davis and Abraham, 2013; Hacker and Pierson, 2014; Vogel, 2012). When the Republicans took control of both houses of Congress in 1994, party politicians claimed this was a signal that Americans expected Congress to ‘tame’ the ‘regulatory beast’ and to stamp out government interference with ‘decisions that are best left to the individual’ (Vogel, 2012: 228), which we understand as part of a neoliberal position (Nik-Khah, 2014).
Whether or not successive Republican majorities in both houses of Congress genuinely reflected a more ‘anti-government’ national mood, it is possible that their political rhetoric was effective in reshaping the FDA’s institutional culture. This might have occurred through the political appointment of senior agency officials who shared a neoliberal ideology and worked to change agency practices and norms in that direction. Or it may have occurred because FDA staff believed the changing political environment signaled a shift in broader cultural values, including a preference that the FDA adopt a less paternalistic, more consumerist approach. In either scenario, the FDA’s probing scientific culture might come to co-exist with a more consumer-oriented institutional culture, such that agency staff increasingly came to see themselves as ‘information providers’ whose role was to facilitate patient-clinician decision-making and choice in the ‘medical marketplace’, rather than as gatekeepers whose role was to protect the public from unsafe or ineffective drugs.
Here again, there may be overlaps between this explanatory framework and other theories discussed above. For example, the claim that political appointees who championed a more business-friendly, neoliberal approach were partly responsible for reshaping FDA culture is consistent with corporate bias theory (Davis and Abraham, 2013). Similarly, increasing patient activism from the 1980s onwards (‘disease-politics’) may have encouraged FDA scientists to see patients as ‘consumers’ who had the right and were willing to take risks and exercise choice with respect to treatments. It is important to note that a neoliberal approach to medicines regulation would not necessarily entail the existence of disease-politics, since physicians would also be seen as consumers in the medical marketplace. Nevertheless, disease-politics would tend to strengthen and confirm a more market-oriented approach.
Methodology
We collected and reviewed in-depth a range of documentary evidence to construct as complete a picture as possible of the chain of events, interactions and decision-making processes that led to the approval of Relenza. These included publicly available agency documents: the FDA’s medical and statistical reviews, advisory committee meeting transcripts, memos from meetings and discussions between the manufacturer and FDA personnel, and approved labels. The numerous and detailed memos, covering the entire process from submission to approval, offered a ‘real time’ perspective impossible to obtain from reading only the regulatory reviews (in part written retrospectively), or by relying on oral accounts years after the fact. To ensure proper understanding of the clinical research and regulatory contexts we also reviewed the scientific and ‘grey’ literature on neuraminidase inhibitors, the European regulatory reviews of Relenza, and a range of media, policy and academic commentaries and research on the FDA during the late 1990s and early 2000s.
We utilize process-tracing as a method of qualitative, within-case analysis to establish whether the events and processes we describe in the case fit those that would be predicted by the distinct, albeit partially overlapping, frameworks outlined above (Bennett and Checkel, 2015). Specifically, we consider the probative value of different pieces of evidence, and sometimes absence of evidence, to affirm or alternatively cast doubt on various explanations. In testing the explanatory strength of alternative accounts, we ‘cast the net widely’ (Bennett and Checkel, 2015: 18) and draw on interpretations offered by the medical and lay press and claims by FDA managers and scientists, as well as theoretical and empirical insights from previous studies of pharmaceutical regulation.
Empirical analysis
DAVP reviews and the negative Advisory Committee vote
In late 1998 GlaxoWellcome (since 2000 GlaxoSmithKline) submitted an NDA for Relenza to the FDA’s Division of Antiviral drug Products (DAVP). The NDA included studies designed to demonstrate the drug’s safety and efficacy. This section begins by discussing the design and results from the three, pivotal trials carried out in Europe (EU), the Southern Hemisphere (SH) (primarily Australia), and North America (NA) (primarily the US), respectively. The primary endpoint across the trials was time to alleviation of influenza symptoms, and – to mimic the real-life use of the drug – patients were allowed to use over-the-counter cold medication at their discretion.
Patient enrollment to studies was based on symptoms of influenza and, subsequently, 71% to 77% of patients were confirmed to have an influenza virus infection. Table 1 shows GlaxoWellcome’s main analyses in the entire trial population (so-called intent-to-treat population) and the influenza-positive sub-population. However, because DAVP considered that results in the influenza-positive population were the most relevant, since this addressed drug activity on the target disease, DAVP’s efficacy review centered on results in influenza-positive patients (FDA, 1999a).
Results on the primary endpoint: Median time to symptom improvement (days) in pivotal Relenza trials.
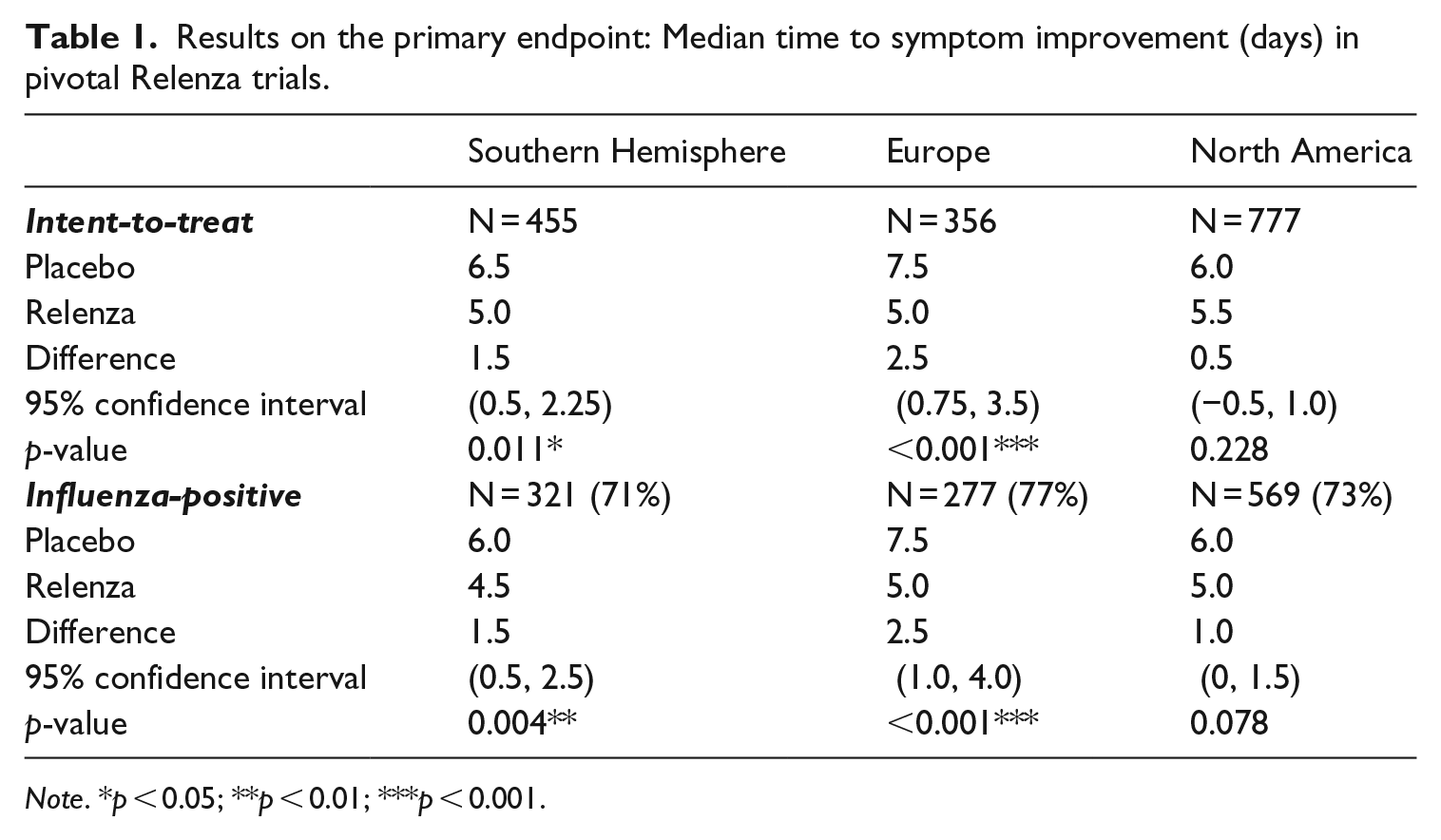
Note. *p < 0.05; **p < 0.01; ***p < 0.001.
Overall, both the DAVP medical officer and statistical reviewer assigned to evaluate the Relenza NDA judged the SH and EU studies to be positive because both trials showed conclusive and relevant therapeutic effects. However, because the NA study had only small and statistically inconclusive point estimates (p = 0.078), despite being well powered, this trial was considered to be negative (FDA 1999a). The implication was that the drug had not been shown efficacious in the US study population.
In an effort to counter the DAVP reviewers’ concerns, GlaxoWellcome suggested, in their exchange with the DAVP, that the NA study should be viewed not as negative but as ‘less positive’ than the other two trials (FDA, 1999b). GlaxoWellcome’s argument was that effects in influenza-positive patients approached statistical significance. However, this argument did not persuade the acting statistics team leader, Dr Aras, who noted that while p = 0.078 might be interpreted as a ‘trend in the right direction’, post-hoc analyses by Dr Elashoff, the DAVP statistical reviewer, using alternative statistical tests or endpoints, showed that this trend was not replicated in most analyses and was therefore not robust (FDA, 1999b). Dr Elashoff’s statistical review summarized this negative view on the NA trial: [The] small treatment effects, lack of significance, and very high power to exclude clinically relevant treatment effects, were all seen over the broad spectrum of endpoints of this study. The US [NA] study therefore, when looking at the combined weight of evidence, is considered to be negative. . .. Further, the results of the foreign studies, while convincing evidence of efficacy in those populations, only heighten the concern over the US [NA] study results. These studies proved that any of variety of analyses of symptoms can find significant differences in favor of zanamivir, if such differences exist. (FDA, 1999c: 13, emphasis in original)
Moreover, Dr Elashoff’s report noted that there was an obvious explanation for the difference in results between studies, namely that use of relief medication was almost twice as high in the negative NA study compared to the clearly positive EU study, with the SH study laying in between. A reasonable conclusion, therefore, was that Relenza did not provide any more benefit than did ordinary cold medication and this explained the discrepant results between trials. According to Elashoff, this lack of proven benefit in the US population was particularly worrying since the drug’s harms were poorly understood, having been studied in fewer than 3000 patients. This meant that, if the drug was approved, potentially millions of people with influenza symptoms would be exposed to largely unknown risks without receiving any major benefits.
The DAVP’s evaluation was presented to an FDA Antiviral Drugs Advisory Committee on February 24, 1999, made up of expert scientists and expert consultants from major US medical centers and public health government institutions. Despite meeting deliberations about pandemic risks and the pressing need for new anti-influenza drugs, there was an overriding concern about Relenza’s limited efficacy, especially in the US population, and the panelists therefore voted 13 to 4 against recommending approval (FDA, 1999d). The negative votes included some of the country’s top influenza experts, including the Chief of the Influenza Branch of the Centers for Disease Control (CDC). Significantly, the fact that threats of future influenza pandemics did not prevail over experts’ efficacy concerns cast doubt on explanations positing that health and security threats dominated and propelled expert thinking and action in the Relenza case (Elbe, 2018).
Constructing an argument for approval
In her review of the Relenza application – which in part was written after the Advisory Committee meeting – the DAVP medical officer concluded: [T]he basic decision on whether the drug is approvable for influenza treatment using currently available information can be argued in either direction, and a rationale can be constructed either for non-approval or for approval. (FDA, 1999a: 140, emphasis added)
Memos from meetings and written communication show that, in the months after the Advisory Committee meeting, various rounds of discussions took place between the DAVP and company representatives regarding issues discussed by the Committee and in DAVP reviews. This section details how some key DAVP staff and managers, in the aftermath of the Advisory Committee meeting, actively participated in constructing an argument for approval that would circumvent critical internal reviews and the negative Committee vote.
From an early stage, DAVP management made it clear that they did not view the Advisory Committee’s non-approval recommendation as sufficient reason for rejecting the drug. Thus, the day after the Committee meeting, James Palmer, GlaxoWellcome’s Vice President for Medical, Regulatory and Product Strategy, called on the DAVP Director Heidi Jolson to discuss the status of the application. In the discussion, Jolson ‘emphasized that the review was still on-going and that we [DAVP] consider the role of the [Committee] as “advisory” as with any other application’ (FDA, 1999e).
But despite the DAVP Director’s assurances, in the ensuing days GlaxoWellcome attempted to pressure the DAVP into disregarding the Advisory Committee guidance. Specifically, GlaxoWellcome sent a lengthy letter on March 2, signed by James Palmer and addressed to DAVP Director Jolson, officially complaining, amongst other things, that the DAVP position on Relenza ‘is completely at odds with the will of Congress that drug development and approval proceed swiftly and surely on the basis of reliable communication between sponsors [pharmaceutical companies] and FDA’ (Willman, 2000). The letter also accused the DAVP of ‘blindsiding’ the company during the review process, claimed that the DAVP medical officer ‘exerted considerable and, we believe, misguided and inappropriate influence on the review’, called Dr Elashoff’s statistical analysis ‘extreme’, and complained that the ‘advisory process was distinctly biased against fair and open consideration of [Relenza]’ (Willman, 2000).
GlaxoWellcome’s reference to ‘the will of Congress’ was an allusion to provisions of the 1997 FDA Modernization Act (FDAMA) that codified existing FDA initiatives to expedite the drug review process. Consistent with corporate bias theories of regulation, Hilts (2003) and Davis and Abraham (2013) have argued that FDAMA was the outcome of efforts by Republicans in Congress, allied with industry-supported neoliberal think tanks, to impose a more industry-friendly agency culture. It appears GlaxoWellcome sought to exploit the political sentiment, codified in FDAMA, that FDA should work cooperatively with industry, and not delay drug development and approval through imposition of excessively rigid or burdensome standards.
In response to the letter, on March 5, new discussions were held between DAVP and the company, during which the DAVP reassured GlaxoWellcome that it did ‘not expect to take this application to another Advisory Committee meeting’ (FDA, 1999f: 3). Subsequently, on April 1, another meeting was convened to discuss issues raised in letters, faxes and telephone conversations over the preceding month in which the company continued to express disquiet, particularly in relation to DAVP requests for additional analyses of the Relenza trials. Importantly, in addressing GlaxoWellcome concerns, DAVP management explained that they were not asking GlaxoWellcome to submit further analyses ‘in support of non-approval’; rather they framed their requests as ‘offering [GlaxoWellcome] an opportunity to make the best possible case for their application and to participate in trying to construct an argument for approvability’ (FDA, 1999g: 1, emphasis added). However, they also qualified this statement: ‘It is unclear (and not possible to predict) whether such an argument can be successfully constructed but if it can be, input from [GlaxoWellcome] would be necessary.’
To help GlaxoWellcome make a successful case for approval, DAVP staff suggested that company scientists should carry out additional, exploratory analyses of patient outcomes from the NA trial to see whether they could uncover positive results on some relevant, but retrospectively defined outcomes. Such retrospective analyses are commonly referred to as ‘data-dredging’ and are known to be a common source of bias (Boutron et al., 2010). The DAVP’s rationale for soliciting exploratory analyses is clearly instructive of their efforts to assist GlaxoWellcome, and is therefore worth citing in full: It was our impression [on the basis of previous discussions] that these exploratory analyses . . . might show point estimates of treatment effects in NAIA3002 [NA study] more in keeping with the other studies (although still with less impressive p values) than some of the other analyses. We had therefore suggested these analyses as part of the effort to find common ground between NAIA3002 (and North American results in general) and the other studies, as there has been a general concern among reviewers about the lack of convincing treatment effect in NAIA3002. We do consider it important to have a shared understanding that, as discussed previously, it is very difficult to find any convincing treatment effects in NAIA3002, and consider secondary analyses extremely important for support of any marginal effect that can be described in this study. However, there is no intent to try to elicit any analysis that [GlaxoWellcome] is uncomfortable with or believes should not be done. (FDA, 1999g: 3, emphasis added)
In the weeks that followed, GlaxoWellcome submitted a number of novel analyses, including those the DAVP had suggested (FDA, 1999a), explaining: ‘We have tried to be constructive in tone and content in the submissions throughout the month of April’ (FDA, 1999h: 1). Yet the new analyses also failed to show conclusive effects in the NA trial. In fact, they were contradictory, as the DAVP medical officer noted in her review of GlaxoWellcome’s new submissions: The overall pattern emerging from detailed examination of the efficacy analyses for this study is an inconclusive mixture of analyses yielding small differences in favor of zanamivir (generally of questionable clinical or statistical significance), other analyses showing no discernable difference between zanamivir and placebo, and a few outcomes slightly favoring placebo. (FDA, 1999a: 18)
Moreover, in her final analysis, the DAVP medical officer acknowledged that the many post-hoc analyses conducted made any ‘significant’ findings at this point unreliable because of multiple hypothesis testing (1999a: 119). After exhaustive analyses the conclusions regarding the negative study results remained unchanged.
The failure to show convincing effects in the US population, despite the large study size, arguably placed the DAVP in a difficult situation if they wanted to push for approval. The DAVP’s solution to this problem was to acknowledge that the evidence to support approval was slim, but to then characterize the NA study as ‘inconclusive’ rather than negative (FDA, 1999i: 3). Specifically, in constructing an argument for approval, the DAVP Division Director claimed that the NA study, while not demonstrating a positive effect in the US population, conversely, did not ‘negate the robust demonstration of efficacy’ in the other studies (FDA, 1999i: 3). In the DAVP management’s opinion, results from the NA study were, therefore, compatible with the evidence of efficacy in the other studies. While technically truthful, it would be equally correct to say that the study was ‘compatible’ with a zero effect in the US population, or even with a minor negative effect (since the 95% confidence interval did not exclude zero, and in some analyses the drug performed worse than placebo). Arguably, the DAVP’s line of argumentation could be viewed as an attempt to ‘spin’ the data from the NA study to justify approval.
The DAVP advanced three further arguments to justify approval. First, the Division Director wrote that ‘it may be difficult to show convincing effect in an otherwise healthy population with an acute, self-limited illness’, especially in a situation where there is ‘minimization of any treatment effect by use of symptomatic relief medication’ (FDA, 1999i: 2). The obvious counterarguments to this – of which FDA managers were fully cognizant, since they had been made in earlier DAVP reviews (see above) – were: (1) the smaller EU and SH studies demonstrated significant (albeit clinically unimpressive) differences, which suggest that effects could be shown if they existed, and (2) if treatment effects were minimized by relief medication, there was limited rationale for using Relenza instead of cheaper over-the-counter medication with better defined side-effects. Indeed, the DAVP Division Director acknowledged that although Relenza seemed safe in healthy individuals there was one ‘potentially significant safety concern’ (FDA, 1999i: 7) related to reports of bronchospasm and/or decline in lung function in some patients with underlying respiratory disease.
Second, the DAVP suggested that the FDA should be consistent in its approach (1999a: 152) and referred to the fact that the agency had previously approved two influenza drugs, rimantidine and amantadine, on the basis of slim evidence, allegedly because of difficulties in designing studies that could demonstrate efficacy. The DAVP Division Director cited the FDA’s positive evaluation of rimantadine in the 1980s, which was based on results from 126 patients receiving rimantadine, and where ‘the data was not strong’ (FDA, 1999i: 2). In other words, the DAVP justified its permissive approach by reference to regulatory decisions made more than a decade earlier on the basis of outdated regulatory standards. Moreover, the Division Director simultaneously acknowledged that the regulatory environment and regulatory standards had changed, noting that ‘the zanamivir application contains larger, more uniformly conducted studies and more prospectively defined analyses than those submitted in support of either rimantidine’s or amantidine’s efficacy . . .’ (FDA, 1999i: 2).
Third, the Division Director referred to ‘the public health need’ for new anti-influenza drugs because of the risk of viral resistance developing against the available drugs (FDA, 1999a: 152). However, according to FDA’s expert advisors this argument was problematic because the need was for efficacious alternatives and Relenza efficacy seemed limited at best. Indeed, the DAVP’s review had noted that evidence of efficacy and safety was especially limited in those patients at greater risk of a more prolonged and/or severe course of illness (the elderly and patients with an underlying medical condition), and that the drug had not been shown to reduce complications of, or deaths from, influenza. Consequently, the FDA refused to recommend the drug for ‘high-risk’ patients and restricted approval to treatment of ‘uncomplicated acute illness’ (Mulinari and Davis, 2017). Together, these judgments tend to undermine the argument for Relenza as a ‘public health’ drug.
‘Trying out’ ambiguous and ‘generous’ label language
While DAVP managers satisfied themselves they had constructed an argument for approval, there was still the problem of how to describe the drug’s equivocal study results in the product label consistent with FDA’s requirement that product labels should convey effects accurately and with relevance to US doctors and patients. This section details how the DAVP strategically developed label language that might justify marketing and use of the drug.
Even prior to the Advisory Committee vote, the DAVP had identified as a major issue the question of how to describe the negative NA study in the label. Thus, the medical officer’s report states that suggestions regarding labeling were conveyed to GlaxoWellcome on two occasions before the Advisory Committee meeting (FDA, 1999a: 149), and a memo from three weeks before the Advisory Committee vote cited Dr Elashoff explaining the difficulty faced by the agency ‘in how to construct label wording that would represent the [NA] study results’, to which GlaxoWellcome had replied that they ‘also see the issue as how to arrive at appropriate label descriptions.’ (FDA, 1999j: 2) The memo also cited DAVP staff saying they were ‘troubled by implications for describing treatment effects if it can be dampened out by standard over-the-counter (OTC) drugs’ (FDA, 1999j: 1).
After the Advisory Committee vote, labeling discussions intensified. DAVP staff focused on finding ways to circumvent the negative opinions and recommendations by communicating reservations about efficacy in the label. Thus, a May 14 memo describes a DAVP/company teleconference during which the DAVP outlined ‘some steps we are taking to try to further a shared constructive approach’ regarding Relenza (FDA, 1999h: 2). The DAVP noted that they ‘continued to be concerned by the lack of convincing demonstration of treatment effect in the largest phase 3 study [NA] . . . and by the difficulties of deriving a meaningful generalizable description of treatment effect . . .’ (FDA, 1999h: 2). Nonetheless, the DAVP indicated they had ‘been working on label comments attempting to try out how current understanding of treatment effect could be incorporated into label language’ (FDA, 1999h: 2, emphasis added), and that they would communicate suggestions to GlaxoWellcome for comment in due course.
Over the following month, label language appears to have dominated the agenda leading up to a key meeting on July 1 that would deal with outstanding issues. Accordingly, a June 25 memo describes GlaxoWellcome and DAVP discussions on the draft agenda for that July 1 meeting, and cites DAVP staff commenting: ‘As the applicant is aware, of the various issues of concern to [DAVP], achieving an appropriate description of treatment effects as they become apparent in the review is among the major ones’ (FDA, 1999k: 2).
Then, on June 29, DAVP Division Director Jolson received a call from Palmer, GlaxoWellcome’s Vice President for Medical, Regulatory and Product Strategy, in which he communicated that ‘[i]ncorporation of the numerical data from all three trials [i.e. NA, SH and EU] is of primary importance to the company’ (FDA, 1999l). The reason for GlaxoWellcome’s insistence on having data from all studies in the label is not specified, but the memo notes that Palmer requested permission to include marketing representatives at the meeting, which was granted. It is not unreasonable to assume that this was ‘of primary importance to the company’ because GlaxoWellcome wanted to cite treatment effects of 1–2.5 days in Relenza marketing material based on incorporation of these figures in the label (a claim that European regulators had authorised; see Mulinari and Davis, 2017).
The key July 1 meeting to discuss approval and outstanding labeling issues included 30 FDA staff and 13 GlaxoWellcome representatives (FDA, 1999m). The DAVP Division Director Jolson stated in the meeting that while the FDA judged the company to have ‘done a fine job with development of zanamivir . . . the actual study results are such that arguments against approvability can be made’ (FDA, 1999m: 2). In particular, the fact that ‘two studies show activity but the third is inconclusive and does not show a positive result’ made it ‘a dilemma to try to determine a treatment effect generalizable to a North American population, and it is suspected that any such effect is small.’ Yet, despite this acknowledgement that nothing had changed in terms of the underlying evidence, Jolson indicated that approval was possible, on condition that the label made clear ‘the smallness of the benefit and information about who may and may not benefit’ (FDA, 1999m: 3).
But how should they convey in labeling, without undermining the justification for approving the drug, that the largest trial – and the only one recruiting US patients – was negative? The medical officer’s report clearly captured the difficulty of the task: The one-day point estimate for median difference . . . was at the lower margin of difference that might be considered clinically meaningful; furthermore, with a p value above .05 despite a well-powered, overenrolled study . . . and without strong support from secondary analyses, it could not even be said that the study ‘showed’ a one-day difference in outcomes. Some subgroups showed smaller or even negative point estimates . . .. (FDA, 1999a: 144)
The DAVP’s proposed solution was to include a statement that the NA study – together with a smaller NA phase II study – ‘suggested’ a treatment effect of ‘up to one day’ although results were not statistically significant. Notably, this was consistent with the DAVP’s technical justification for approval, that is, that results were compatible with a treatment effect in the US population. The DAVP frankly described this solution as ‘an attempt at the most generous statement justifiable taking into account phase 3 and phase 2 studies, primary and secondary endpoints, and lack of statistical consistency’ (FDA, 1999m: 5, emphasis added). To this, GlaxoWellcome’s Palmer responded that they wanted to have ‘the drug available for the 1999 influenza season’ and ‘would agree to caveats/qualifiers in the label’ (FDA, 1999m: 3). However, the company expressed dissatisfaction with the ‘up to one day’ wording. As they had made clear, they wanted to include results from non-NA studies too; preferably they wanted to present results as a range of expected effects, that is, 1–2.5 days (FDA, 1999m: 6).
It is significant that the DAVP rejected GlaxoWellcome’s request on this latter point because it demonstrates that DAVP preserved a degree of independence from the company. In particular, the DAVP drew attention to the fact that the EU study was uniquely isolated at the particularly high effect estimate. GlaxoWellcome appears to have accepted DAVP’s view on this because they were quoted saying that they too ‘consider the EU study to be an outlier’ (FDA, 1999m: 6). Nevertheless (and possibly as a compromise) the DAVP suggested mentioning results from SH and EU studies in the label, ‘along with some possible contributors to the difference’, that is, use of relief medication, but not in such a way that ‘it might be mistakenly interpreted as generalizable to the target [US] population’ (FDA, 1999m: 5). In response, GlaxoWellcome said they were still ‘uncomfortable’ because they couldn’t ‘explain to their other constituencies how “up to one day” was derived’ (FDA, 1999m: 6). This statement suggests that GlaxoWellcome saw the ‘up to one day’ interpretation of the NA study – framed by DAVP ‘as the most generous statement justifiable’ – as being far from self-explanatory. DAVP staff responded that the ‘only alternative might be a general statement such as that a slight effect was observed’, and acknowledged, somewhat contradictorily, that ‘a simple clear statement may not be achievable for a guide to physicians prescribing in North America, as there is not a clear quantifiable effect applicable to this population after exhaustive analyses (nor after the initial primary analysis alone)’ (FDA, 1999m: 6).
Following these negotiations, on July 26, the FDA approved Relenza without soliciting any further Advisory Committee input, and with the following label description of the main efficacy results (FDA, 1999n): A phase 2 and a phase 3 study conducted in North America (total of over 600 influenza-positive patients) suggested up to one day of shortening of median time to this defined improvement in symptoms in patients receiving zanamivir compared with placebo, although statistical significance was not reached in either of these studies. In a study conducted in the Southern Hemisphere (321 influenza-positive patients), a 1.5-day difference in median time to symptom improvement was observed. Additional evidence of efficacy was provided by the European study.
In an attempt to explain possible contributors to the difference between studies, the following statement was included: The magnitude of treatment effect varied between studies, with possible relationships to population-related factors including amount of symptomatic relief medication used.
Discussion
Permissive regulation and the strategic use of labeling
The FDA’s approval and labeling of Relenza can be characterized as an instance of permissive regulation in that the FDA deviated from its own scientific and regulatory standards. First, after DAVP reviewers concluded that no convincing treatment effects in the US study population had been demonstrated, senior managers encouraged the company to undertake post-hoc, exploratory analyses to strengthen the case for approval. This was despite a clear understanding that this kind of data-dredging raises the problem of multiple testing, which in turn increases the risk of a false positive. Second, when these additional analyses failed to yield the desired results, DAVP managers claimed that although the results were not statistically significant this did not negate a benefit in the US population – a form of argumentation that is equivalent to ‘spinning’ the data since the NA study had failed to disprove the null hypothesis of no treatment effect. Moreover, in advancing this interpretation of results, DAVP managers effectively ignored DAVP’s own statistical analyses. These analyses undermined the company’s position that the results ‘approached’ statistical significance, since they showed estimates from the NA trial to be neither compelling nor robust. Indeed, additional analyses showed that in some key subgroups, placebo numerically outperformed Relenza.
DAVP managers also attempted to ‘explain away’ the negative NA study by pointing to the difficulty of demonstrating drug efficacy in influenza. They argued that the historical bar for approval of anti-influenza drugs had been set low, so a lower standard for approval should be applied to Relenza. The implication of this argument is that regulation need not keep pace with advances in science or clinical trials methodology if this would be unfair to companies. This position is clearly contrary to FDA’s stated mission to protect and promote public health, and is contradicted by the historical reality, which saw the FDA take a leading role in advancing the science of drug development and testing (Abraham, 1995; Carpenter, 2010).
The FDA’s approval of the Relenza label can also be characterized as permissive. Once a decision to grant market approval for Relenza had been made, DAVP staff and management strategically crafted and negotiated a label that included ‘the most generous statement justifiable’, suggesting Relenza might be efficacious in the US population despite the statistically non-significant results. While the DAVP did not capitulate to the company’s request to present data from all three pivotal studies in the label, ambiguous label language, describing Relenza’s effect in the NA study, failed to meet regulatory standards that the label convey clear, informative and accurate prescriber-relevant information (FDA, 2006).
But problems with the DAVP’s construction of label descriptions do not end there. As we have seen, one condition for approval was that the label should appropriately represent the magnitude and the level of certainty of the treatment effect in the ‘target population that would be likely to receive the drug’ (FDA, 1999m: 3). Crucially – since there was no (and still is no) sufficiently rapid ‘point of care’ influenza diagnostic test – the target population for Relenza were individuals with influenza symptoms (the intent-to-treat population), not those with laboratory-confirmed disease (the influenza-positive population). This is important because the large majority of patients, possibly around 85%, eligible for the drug in most settings will not in fact have influenza, but instead be infected by one of the many agents that produce overlapping ‘influenza-like symptoms’ (Jefferson et al., 2014). Yet, had the label reported trial results for the intent-to-treat population this would have reduced the apparent treatment effect in the NA study to an even less compelling half-a-day reduction in duration of symptoms (p = 0.228; 95%CI, –0.5, 1.0) (see Table 1). This would have made it even more difficult to justify approval since DAVP maintained that a one-day improvement was at the lower limit of clinical relevance (FDA, 1999a: 23).
Interrogating alternative explanations for FDA’s actions
In the following section we consider the extent to which both social science and official accounts of US pharmaceutical regulation might explain the FDA’s permissive approval and labeling of Relenza. Consistent with a public interest theory of regulation, the DAVP itself attempted to provide a public health justification by pointing to the upcoming influenza season and the need for effective drugs to treat this disease. However, the evidence we have reviewed does not support the claim that FDA genuinely believed Relenza to be a public health advance since it is clear from our analysis that the agency had very low expectations of drug efficacy. In particular, FDA did not recommend use of the drug by patients most at risk of the complications of influenza due to subsequent infections – that is, precisely those patients with the greatest health needs (Mulinari and Davis, 2017).
A related possibility entertained by Elbe (2018) is that DAVP managers approved Relenza because they were influenced by expert concerns over pandemic health and security threats. However, we have argued that the notion that these concerns dominated FDA’s thinking is not supported. First, it is inconsistent with the fact that the FDA’s Advisory Committee for Relenza – comprised of key government and academic researchers in epidemiology and public health, infectious disease and clinical medicine – voted overwhelmingly against approval, despite deliberations about pandemic risks and the need for new anti-influenza drugs. Second, the FDA only approved the drug for symptomatic relief of uncomplicated influenza in otherwise healthy adults and not as a prophylactic ‘countermeasure’ to stop viral spread in epidemic or pandemic situations – casting further doubt on the proposition that fears of a looming pandemic underpinned DAVP decision-making. It is difficult, therefore, on the basis of the available evidence, to construct a plausible case that the primary driver of FDA’s permissive regulation was the conviction that, by approving Relenza, the agency would be fulfilling its public health protection role.
Another hypothesis is that the DAVP’s actions reflected an increasingly consumerist and neoliberal logic at FDA, as seen, for example, in the agency’s move to legalize direct-to-consumer advertising (Donohue, 2006). Such a regulatory logic might involve a reframing of the notion of the ‘public interest’, such that a key regulatory goal would be facilitating maximum consumer choice and supporting physician and patient decision-making. At first glance, there appears to be some evidence to support this thesis, both from previous studies of the FDA and in the Relenza case. In a study of FDA’s implementation of risk management as a regulatory tool for managing severe drug risks, Davis and Abraham (2011, 2013) show that between 1995 and 2005, the FDA were more likely than EU regulators to warn physicians about drug risks rather than withdraw unsafe drugs. Moreover, Davis and Abraham report that senior FDA managers cited ‘doctor and patient autonomy to choose their own risks as justifications for keeping such drugs on the market’ (2011: 427). It is possible, therefore, that at the time of Relenza’s approval, the FDA’s institutional culture was increasingly influenced by more extreme, consumerist values – although there is also evidence that such views were not uniformly embraced throughout the organization (Davis and Abraham, 2011: 426).
Similarly, in the Relenza case, DAVP managers argued that approval of Relenza could be justified if the label conveyed information on the smallness of the drug’s benefit and information about who may and may not benefit (FDA, 1999m: 3). However, the thesis that FDA officials were primarily motivated by a desire to facilitate individual physician and patient decision-making becomes less compelling when we consider this would entail that regulators design informative drug labels that could be readily understood. The opposite occurred in the case of Relenza, with DAVP resorting to the use of vague, ambiguous and non-informative language to describe Relenza’s unproven benefits in the US population.
We would suggest, therefore, that to the extent that various constructions of ‘the public interest’ or a distinctly consumerist and neoliberal logic played a part in the FDA’s regulation of Relenza, this was not because they were primary drivers of FDA’s decision-making but because they provided managers with a convenient rhetoric to rationalize their actions.
Building on reputational theories one might hypothesize that DAVP’s actions were driven by a wish to bolster FDA’s reputation with one or more of the agency’s ‘audiences’. Most clearly perhaps, DAVP’s official approval justification suggests it wanted to uphold FDA’s reputation for prioritizing public health. At the same time, however, managers seemed unconcerned with damaging FDA’s reputation for rigor with its science audience, mainly academics and medical professionals inside and outside the agency, including members of its Advisory Committee, which does not fit well with previous reputational accounts of FDA behavior. Importantly, Moffitt (2010) has suggested that the FDA uses Advisory Committees primarily to present a favorable external agency image and to preempt subsequent criticism. But with Relenza, FDA officials risked damage to the agency’s public image by ignoring its advisors and, indeed, the approval of Relenza became a point of media criticism which arguably undermined rather than enhanced the FDA’s reputation for scientific rigor (Frontline, 2003; Willman, 2000). Related, reputational accounts have shown that lobbying by patient groups can influence FDA decisions (Carpenter, 2004), consistent with disease-politics theory, but there is no evidence of major patient activism either in relation to influenza, or in the case of Relenza.
This would suggest, then, that if FDA decision-making were driven by reputational concerns, the agency was not primarily motivated by worries over the views of its scientific audience, nor by worries over the views of patient advocates (since patient activism was absent in this case). However, this does not rule out an explanation drawing on reputational theory. As argued by Carpenter (2010), the relative social and political power of different FDA audiences – and their attendant ability to threaten FDA’s reputation – have shifted over time. And in the mid-to-late 1990s, FDA’s reputation was under attack by the political wing of the US pharmaceutical industry, and by anti-FDA, industry-funded campaigners and think tanks with links to the Republican party. Indeed, plans to ‘reform’ FDA began in earnest within a few short months of the 1994 Congressional election results, which gave Republicans control over Congress (Davis and Abraham, 2013: 72–76).
While some of the more extreme blueprints for FDA reform did not make their way into FDAMA in 1997 (Carpenter, 2010; Davis and Abraham, 2013), by the time of Relenza’s review the FDA had experienced years of intense Congressional scrutiny, during which time powerful Republicans such as Newt Gingrich put forward proposals for the wholesale privatization of the drug review process (Carpenter, 2010: 731; Davis and Abraham, 2013: 72–76). It is plausible that agency officials felt under pressure to approve Relenza to preempt (further) anti-FDA sentiments in Congress, especially after GlaxoWellcome decided to make the FDA’s handling of Relenza a public matter by issuing the high-level letter of complaint to the FDA (see below).
Reputational accounts of FDA have mainly taken the form of meso- and micro-level analyses to account for institutional change and decision-making. However, Carpenter (2010, 2014) acknowledges the importance of the wider macro-political environment of the 1990s, and entertains the possibility that it facilitated a degree of ‘cultural’ capture of the FDA, whereby FDA managers’ attentiveness to the new political climate of the 1990s became encoded and reflected ‘at the level of everyday regulatory practice’ (Carpenter, 2010: 733; see also Kwak, 2014). In other words, some reputational accounts overlap with aspects of capture theory by observing that intense political pressure on FDA to develop a more accommodating and less ‘adversarial’ relationship with industry may have enabled ‘an unhealthy culture of closeness between the FDA and industry’ (Darrow et al., 2017: 2283).
This explanation also has commonalties with corporate bias theory (Davis and Abraham, 2013). According to this view, industry’s growing access to, and influence over the three branches of US government has generated a pro-industry ideology and a form of corporate bias within the state, such that powerful economic interests are systematically favored at the expense of diverging or conflicting interests, such as patient and public health (Davis and Abraham, 2013; see also Hacker, 2006). Linking these macro-level changes with meso- and micro-level analyses of FDA over the period discussed in this article, Davis and Abraham (2013) argue that corporate bias within the state facilitated a degree of industry capture amongst senior FDA management. For the FDA’s rank-and-file scientists, this led to ‘an acculturated resignation towards the futility of challenging pharmaceutical firms, senior managers and FDA advisory committees to ensure that regulatory standards to protect public health were upheld contrary to the commercial interests of industry’ (Davis and Abraham, 2013: 266).
According to corporate bias theory, then, FDA’s permissive regulation in the case of Relenza can be understood in terms of the top-down implementation of a more conciliatory and cooperative approach towards industry in response to political and industry pressures. Indeed, GlaxoWellcome made this pressure on the FDA explicit by referring, in its letter to FDA managers, to ‘the will of Congress that drug development and approval proceed swiftly and surely’ (Willman, 2000) – after which the DAVP began shepherding Relenza towards approval.
Much of the evidence we have reviewed is consistent with a corporate bias theory of regulation, but also with aspects of Carpenter’s (2014) description of a form of ‘cultural’ capture of the FDA. For example, DAVP management’s cooperative attitude is seen in their frank statement – made less than one month after GlaxoWellcome’s letter – that they were ‘offering [GlaxoWellcome] an opportunity . . . to participate in trying to construct an argument for approvability’ (FDA 1999g: 1). Similarly, Dr Elashoff, in response to a journalist’s direct question in 2003 about why the FDA approved Relenza, said: I think most reviewers at FDA . . . that you talk to would find it’s pretty well understood that the large pharmaceutical companies kind of have a substantial influence on the review process. I don’t think anyone is really surprised about that. Someone who would disagree maybe isn’t really involved in reviewing drugs, because I think nearly every application I was involved with or I knew about, where it looked like there were concerns about the drug, it was always understood that there would be high-level complaints from the company to FDA management. Those complaints would come down to the reviewers. There would be both subtle and rather overt pressure to change the reviews, change the recommendations, soften the language. (Frontline, 2003)
Strong support for Elashoff’s claim comes from the fact that, following his critical handling of Relenza, managers removed him from the review of Relenza’s cousin, Tamiflu. Such events may send a message to agency staff, and have consequences in terms of shaping a broader institutional culture of resignation: A lot of the other reviewers at FDA thought they liked the fact that I made a stand on Relenza, but said they wouldn’t have done it themselves, and that it wasn’t going to be good for my career at FDA in the future. So in one sense, it was gratifying to have people say that they agreed with me and liked what I did. On the other hand, it was fairly disheartening to know that no one else is really going to step up and do the same thing on drugs that they were reviewing that had similar problems. (Frontline, 2003)
Further support for Elashoff’s account of these micro-level institutional dynamics comes from three major surveys of FDA personnel, which also suggest that Dr Elashoff’s sentiments were not atypical or specific to the case of Relenza, the DAVP or GlaxoWellcome. Rather, the wider sociopolitical and legislative shifts experienced by the FDA through the late 1990s appear to have had profound and continuing consequences for the internal working environment of the agency. In 1998, the majority of FDA medical reviewers responding to a survey reported experiencing more pressure to approve a greater proportion of new drugs than they had experienced in the period prior to 1995 (Lurie and Wolfe, 1998). The medical officers cited several instances where they had been instructed not to present data or their own negative opinions to an Advisory Committee, or had been asked to change their opinion to agree with supervisors. Similarly, a 2000 Internal FDA Survey of Personnel found that about one third of respondents did not feel comfortable expressing their differing scientific opinions, with some reporting pressure to favor the wishes of manufacturers over the interests of science and public health, and receiving requests from senior agency officials to alter their opinions (FDA, 2000). Finally, in a 2003 Department of Health & Human Services Inspector General Study of FDA, eighteen per cent of surveyed FDA physicians and scientists reported pressure to recommend that drugs be approved despite their reservations about the drug’s safety, efficacy or quality (OIG, 2003).
Industry bias and the interplay of institutional and scientific cultures at the FDA
Returning to the puzzle we posed at the start of this article, how to account for the fact that the FDA approved Relenza in the face of doubts over the drug’s efficacy, we find little or no evidence that the FDA was primarily driven by public health concerns (including wider fears of a looming pandemic), disease-politics, or a consumerist and neoliberal regulatory logic. Of course, it is entirely possible that disease-politics, for example, was an important influence in specific approval decisions throughout the 1990s and 2000s. Or that senior FDA managers, or the institution as a whole, increasingly embraced a consumerist and neoliberal regulatory logic. However, there is little or no evidence that these were significant factors shaping decision-making in the Relenza case – except insofar as they provided the DAVP with palatable justifications for specific regulatory actions.
Instead we find evidence – consistent with some reputational and capture theories, as well as with corporate bias theory – to support the claim that FDA managers responded to political and industry pressures by adopting a conciliatory and cooperative approach to companies. Specifically, we find that the FDA managers may work closely with companies to get drugs approved, despite negative opinions and recommendations from the agency’s internal and external experts, including through suggesting analyses and interpretations that strengthen the case for approval, and through crafting ambiguous statements and generous interpretations of study results to print in the product label. In this respect, our case study analysis adds to a substantial and growing social science and pharmaceutical policy literature that documents drug industry biases at seemingly each and every step along the product lifecycle of prescription drugs – including biases in relation to research and drug development (e.g. Fisher, 2008; Lundh et al., 2017; Sismondo 2009), regulation (e.g. Davis and Abraham, 2013; Homedes and Ugalde, 2014; Nik-Khah, 2014) access, cost and pricing (e.g. Light and Warburton, 2011; Ozieranski and King, 2017; Weissman, 2017) and marketing (e.g. Dumit, 2012; Mulinari, 2016; Schwartz and Woloshin, 2019).
It is important, however, to recognize that the FDA did not entirely capitulate to company demands with respect to product labeling, and frequently challenged GlaxoWellcome’s interpretations of the data. For example, the FDA did not agree to labeling that implied effects seen in EU and SH studies could be directly extrapolated to the US population. And as we documented in a previous article (Mulinari and Davis, 2017), the FDA rejected GlaxoWellcome’s claims about Relenza’s efficacy in high-risk populations and its capacity to reduce potentially life-threating complications of influenza – claims that were accepted European regulators. We explain differences between the US and EU in terms of the FDA’s strong scientific culture which entailed more rigorous and probing review methodologies (Mulinari and Davis, 2017).
As Carpenter (2014) has argued, there are degrees of regulatory capture and it is important that scholars attempt to identify those factors that might operate as protective mechanisms. An important implication from our studies is that FDA’s scientific culture appears to have mitigated, to some extent, the impact of an institutional culture in which industry was increasingly viewed as the FDA’s ‘client’. Our analyses also suggest that ‘capture’ of rank-and-file FDA scientists was relatively weak, and that even amongst senior managers there was a counter-pressure to observe norms of regulatory science. However, institutional and scientific cultures do not exist as distinct entities. Indeed, the FDA’s meticulous review practices were rooted in an older, more adversarial institutional culture that valued and promoted an ‘arms-length’ relationship with industry (Abraham, 1995; Carpenter, 2010). In this sense, our case study of Relenza gives a unique insight into contradictions attendant on an agency in transition. On the one hand, the FDA as an institution had developed a scientific culture that encouraged critical and independent examination of industry claims, strict norms of regulatory science, and rigorous, penetrating review practices. On the other hand, the FDA was under pressure from Congress to facilitate and expedite the review of drugs and adopt a conciliatory and cooperative approach towards industry. With Relenza, senior FDA managers had to reconcile critical medical and especially statistical reviews – the products of an established scientific culture – with these new managerial imperatives, ultimately resulting in permissive regulation.
If correct, this insight may have implications for present-day FDA politics and practice. A major recent topic of debate concerns the FDA’s growing and significant dependence on fees from the firms whose products it regulates, with each successive reauthorization of the user fee legislation requiring FDA to be increasingly responsive to industry (Darrow et al., 2017). And there has been renewed and intense political pressure on the FDA to further accelerate the drug review and approval process (Downing et al., 2017). In this sense, the broader ideological and political climate that formed the backdrop to FDA’s regulation of Relenza persists today.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Swedish Research Council (VR) [Grant 2013-1268 to SM]. CD reports grants from the Economic and Social Research Council, United Kingdom, during the conduct of the study.