
Abstract
Biopharmaceuticals, produced by recombinant DNA technology, are generally more complicated to produce than small molecule drugs. As patents around the development and manufacturing of these biopharmaceuticals expire, biosimilars are being developed as comparable and more affordable alternatives to improve patient access and market competition. This commentary explains what a biosimilar is; it compares and contrasts biosimilar production with that of small molecule, generic, and other biological drugs; and it describes basic principles of the nonclinical development program for monoclonal antibody biosimilars.
Keywords
Biotechnology has enabled the development of protein-based therapies for a variety of serious diseases. Worldwide, millions of patients have benefited from approved biologic products, and the global market for therapeutic monoclonal antibodies (mAbs) was $48 billion in 2010. 16 These biopharmaceuticals treat or prevent severe diseases such as cancer, heart attacks, stroke, multiple sclerosis, diabetes, rheumatoid arthritis and other autoimmune diseases, hepatitis, neurodegenerative diseases, and rare genetic diseases. However, biopharmaceuticals produced by recombinant DNA technology and protein engineering are more expensive to develop and manufacture compared to chemically synthesized small molecule drugs, which may limit access. 6
The first biologic medicines were approved in the 1980s; patent protection for several has already expired, with the majority of mAb products beginning to come off patent in 2015. 21 As these patents expire, biosimilars are being developed as comparable and more affordable alternatives that improve patient access and enhance market competition, thereby contributing to reduced health care costs for insurers and national health care systems. In the European Union (EU), more than a dozen biosimilars have been marketed since 2006—primarily well-characterized simple recombinant growth factors, hormones, and interferons. Infliximab, the first mAb biosimilar, was recently approved in the EU. 13 In the United States, a biosimilar pathway—or 351(k) package—was created as part of the 2009 Biologics Price Competition and Innovation Act; to date, no biosimilars have been approved in the United States via this pathway.
Biologics: What Are They and How Do They Work?
Biopharmaceuticals (“biologics”) represent a variety of administered proteins, including hormones (eg, growth hormone, insulin, and erythropoietin), enzymes (replacement therapy for those that are naturally produced in the human body), immunoglobulin-based mAbs, blood products (eg, clotting factors), gene or cell therapy products, and vaccines. Like all medicines, biologics work by interacting with the body to produce a therapeutic outcome, but the mechanisms by which they do this can vary from product to product or across therapeutic indications. In general, biologics act on a specific cellular pathway or target and can be used treat more than one disease indication if the underlying mechanism of action/disease pathogenesis is the same.
Biologics are typically larger and more complex than the chemicals that make up small molecule drugs; for example, an IgG1 mAb is approximately 150 000 Daltons compared to morphine at 285 Daltons. While some biologics may be administered in their native form, they are frequently modified to improve pharmacokinetics or alter biologic function. Examples of modifications include changes in specific amino acids to alter specific biologic effector functions (antibody-dependent cellular cytotoxicity), attachment of a specific toxin (antibody-drug conjugates) for increased potency, or chemical additions (eg, polyethylene glycol) to improve circulating half-life of the biologic. However, changes in native conformation also increase the risk of an immune response if the altered protein is perceived as foreign by the body. 22
How Are Biologics Produced?
Biopharmaceutical development uses living systems (plant or animal cells, bacteria, virus, and yeast) and modern DNA and protein engineering technologies to produce biologic medicines that treat human diseases and genetic disorders. Many biopharmaceuticals use genetically modified cells to increase protein expression and the amount of the final drug product. 5,6
Each biopharmaceutical manufacturer has developed unique cell lines designed to produce the desired protein attributes and a proprietary process to produce the protein product at manufacturing scale (typically 20 000 liters). The production of biopharmaceuticals, particularly mAbs, involves complex engineering processes such as fermentation and purification. 4 The manufacturing process for biologics is very sensitive and must be precisely controlled to obtain consistent results and to guarantee the safety and efficacy of the final product. Small changes in the process can result in altered product characteristics, even under well-controlled manufacturing processes. 3,31,34 Analytic tools—such as chromatography (ion exchange, size exclusion, and reverse phase), mass spectroscopy, and capillary electrophoresis—are used to monitor possible variations of biologic products, and a high degree of process control is required to ensure product quality and consistency. 3,16,17
How Do Biologics Differ From Small Molecule Drugs?
Biopharmaceuticals differ in many ways from small molecule drugs, including their molecular size, complexity, and route of administration (Table 1). While small molecule drugs are frequently delivered via oral administration, biopharmaceuticals are typically administered parenterally because their molecular properties (molecular size, hydrophobicity, and gastric degradation) preclude oral administration. 32
Biopharmaceuticals Are Larger and More Complex Than Traditional Small Molecule Drugs.

Small molecules are typically manufactured by well-defined routes of chemical synthesis and can usually be analyzed to determine all the various components. In contrast, most biopharmaceuticals are produced in living systems genetically modified to produce a specific protein in large quantities and then purified through a complex manufacturing process. The complexity of the biologic molecules, as well as the production process, results in heterogeneity in some parameters, such as charge variants (including glycosylation and deamination) and size variants (including aggregates and incomplete formation of disulfide bridges), even between batches. 8,16 Therefore, biopharmaceuticals consist of mixtures of many different forms of the same protein. For example, the 527 amino acid tissue plasminogen activator protein, with 17 disulphide bridges and 3 glycosylation sites, contains more than 1 billion chemically distinct active ingredients in the final drug product. In contrast, typically 95% of the active ingredient molecules in a small molecule drug are chemically identical. 19 This inherent variability of biopharmaceuticals is tightly controlled and monitored by manufacturers and regulators to remain within accepted and predefined limits. 16,17
Immunogenicity
Generally, small molecules are too small to be recognized by the immune system. In contrast, biopharmaceuticals—due to their composition and large molecular size and particularly if they have been modified from their native proteins state—have the potential to be recognized by the body as “foreign” and therefore have the risk of inducing an unwanted immune response.
Many factors contribute to the ability of a biopharmaceutical to induce antidrug antibodies (ADAs), including protein sequence (including similarity to endogenous proteins), posttranslational modifications (glycosylation, oxidation, etc), and higher-order structures such as aggregation. 27 Dose, route of administration, formulation, production process, process impurities (including host cell proteins), and underlying disease state can also influence the development of ADAs. 32
The immunogenic potential of a biopharmaceutical can be both an asset and a liability. Vaccines specifically exploit their immunogenic potential by provoking an immune response that recognizes a foreign organism or substance. However, for other protein-based therapeutics, stimulating an immune response is undesirable since it can result in decreased overall exposure to the therapeutic, thereby reducing overall efficacy, and/or it can lead to adverse effects such as immune complex deposition and complement activation. For example, neutralizing ADAs can block the activity of endogenous proteins, as seen with ADAs to recombinant erythropoietin and factor VIII. 32
In general, human biopharmaceuticals are more immunogenic in nonclinical species, such as rat, dog, and nonhuman primate, due to species differences in protein structure and an expected immune reaction to the foreign human protein in animals. Therefore, while nonclinical ADAs are not predictive of human ADAs, they can be useful in the overall interpretation of nonclinical toxicity data, especially when changes in pharmacokinetic or pharmacodynamic parameters are observed. 22,26
What Are Biosimilars?
A biosimilar is a biopharmaceutical that is purposefully developed to be highly similar to another biologic product (referred to as the reference product) that has already been approved for human administration. The biosimilar must have the same amino acid sequence and the same safety and efficacy profile as the reference product. (If it has improved safety or efficacy, it is not a biosimilar; it is a “biobetter” with a more traditional drug development pathway to approval and marketing. 33 )
A biosimilar has the same dose and route of administration and treats the same disease indications as the reference product but is produced by a different manufacturer, using a different cell line and manufacturing process. Consequently, it is not an exact copy of an off-patent marketed biopharmaceutical. 29 However, it is a very close physiochemical copy with highly similar biological effects. 22
Reverse engineering a marketed biopharmaceutical to make a biosimilar is a more complex process than synthesizing a generic version of a small molecule drug, for which the chemical structure can be copied exactly (Fig. 1). In addition, the manufacturing process for a biosimilar will be different (newer and more economical) and may use a different cell line than the reference product, 33 so some differences between the reference product and biosimilar are expected. However, biosimilars are expected to have the same amino acid sequence as the reference product. It is currently unclear how similar a biosimilar must be to the reference product and what assays are expected to demonstrate equivalent physiochemical properties and biologic effect. 32 Biosimilars are systematically developed to be highly similar to the reference product and are produced to the same manufacturing standards as other biologic products. Any differences between the biosimilar and reference product cannot affect the quality, safety, and efficacy of the biosimilar in humans. 33,34

Biologics and biosimilars are more complex and difficult to develop and manufacture than small molecule drugs. The chemical structure of small molecule drugs can be copied exactly; analytical studies to demonstrate quality and comparable pharmacokinetics (PK) are all that are typically required for approval of the generic version. In contrast, while the biosimilar is required to have the same primary amino acid sequence as the novel biologic, secondary structures (eg, glycosylation) differ, and the molecules are therefore not identical. Comparative analytical, nonclinical, and clinical data are required for approval of a biosimilar.
Potential Economic Benefits of Biosimilars
Biopharmaceuticals are generally more expensive than small molecule drugs and are critical to treating a variety of serious and debilitating diseases. Like the original reference products, biosimilars are generally more difficult and more expensive to develop than small molecule drugs.
The financial impact of biopharmaceutical treatments have been growing over the years, and managing prescribing and reimbursement is challenging for payers such as patients, insurers, and government health care programs. Biosimilars offer a potentially comparable and more affordable alternative to existing biologic medicines that have lost patent protections; they can also enhance market competition, as recently seen with the introduction of trastuzumab biosimilars in India. 11 As a result, the availability of biosimilars may improve access to biopharmaceuticals for more patients and contribute to the financial stability of healthcare systems. Thus, their availability offers potential economic benefit to health care systems while providing global access to the new treatment options for patients brought about by advances in medical science and process engineering. 20
Biosimilar Regulatory Pathways and Guidelines
In 2004, the EU was the first region in the world to establish a legal framework and regulatory pathway for biosimilars, with the first biosimilar medicine approved by the European Medicinal Authority (EMA) in 2006. 9,10 The World Health Organization also issued biosimilar guidance, 35 which shared scientific principles with the EMA guidance and served as a “roadmap” for guidance documents being developed by other countries, including Australia, Canada, Japan, Turkey, Singapore, South Africa, and Taiwan. In 2009, the US Biologics Price Competition and Innovation Act was passed, and 3 biosimilar guidances were released in 2012. 15
In the EU, a biosimilar is a copy version of an already authorized biotherapeutic with demonstrated similarity in physiochemical characteristics, efficacy, and safety, based on a comprehensive comparability exercise. 9,10 The EU defines “the reference product” as the product authorized in the EU, while the US Biologics Price Competition and Innovation Act created an abbreviated approval pathway for biologic products that are demonstrated to be biosimilar to a Food and Drug Administration (FDA)–licensed biologic product. For some biopharmaceuticals, different drug products are approved in different geographies, complicating the global development of biosimilars. For example, the anti-CD20 mAb rituximab is marketed in the EU as MabThera and in the United States as Rituxan. Regulatory guidelines in the United States and the EU both require comparative studies between the proposed biosimilar and the reference product, but different regulatory agencies recognize different reference products. Therefore, to support a global development program for a rituximab biosimilar, a 3-way analytic and clinical pharmacokinetics comparison (MabThera ↔ Rituxan, MabThera ↔ rituximab biosimilar, and Rituxan ↔ rituximab biosimilar) is required to establish a bridge across the 3 molecules. If other countries with biosimilar pathways have the same requirements to use the specific product approved for marketing in their regions as the reference product, the resulting analytical, nonclinical, and clinical studies become overwhelmingly complex and inefficient in terms of cost and animal usage for biopharmaceutical companies attempting a global drug development program. Recognizing this challenge, the EU has recently indicated a willingness to accept reference products authorized outside the European Economic Area if authorized by a regulatory authority with similar scientific and regulatory standards as the EMA. 14
Comparison of the reference product to the biosimilar is a core principle of biosimilar development and a key principle of the analytical characterization, nonclinical in vivo studies, and clinical development program. The scientific principles that underlie the comparability exercise when changes are made in the manufacturing of a given biologic product are the same for the development of a biosimilar. 34 However, data requirements for a biosimilar are higher than those to evaluate a routine manufacturing process change and always include clinical studies. 33
Nonclinical Biosimilar Drug Development: An Overview
The abbreviated development pathway for biosimilars streamlines drug development, reducing development costs, time, and animal usage. The nonclinical development of a biosimilar begins with the selection of the appropriate reference product (or products), the identification of the key molecular and quality product attributes (influencing both safety and efficacy), and understanding the range of variability at release and throughout its shelf life. These features affect everything from clone selection to the final manufacturing process and the development specifications for the biosimilar.
Biosimilar drug development is based on a robust head-to-head comparison of the biosimilar to the reference product in terms of quality, safety, and efficacy. These 3 elements are pursued in a parallel but overlapping fashion:
Quality: comparative analytical and functional characterization of the biosimilar and reference product (continues as an iterative process throughout the biosimilar development).
Nonclinical: comparative nonclinical in vivo studies to rule out any “new” toxicity findings and ensure safe human exposure.
Clinical: comparative clinical studies in a sensitive patient population to demonstrate that any minor differences detected during the quality evaluation are not biologically meaningful and to detect any differences in clinical safety or efficacy.
Compared to the development of novel biopharmaceuticals—that is, the section 351(a) biologics license application in the United States—the section 351(k) biosimilar license application pathway has a more extensive analytical and functional characterization component. In addition, the nonclinical and clinical components of the biosimilar licensing application are typically reduced compared to novel biopharmaceuticals (Fig. 2).
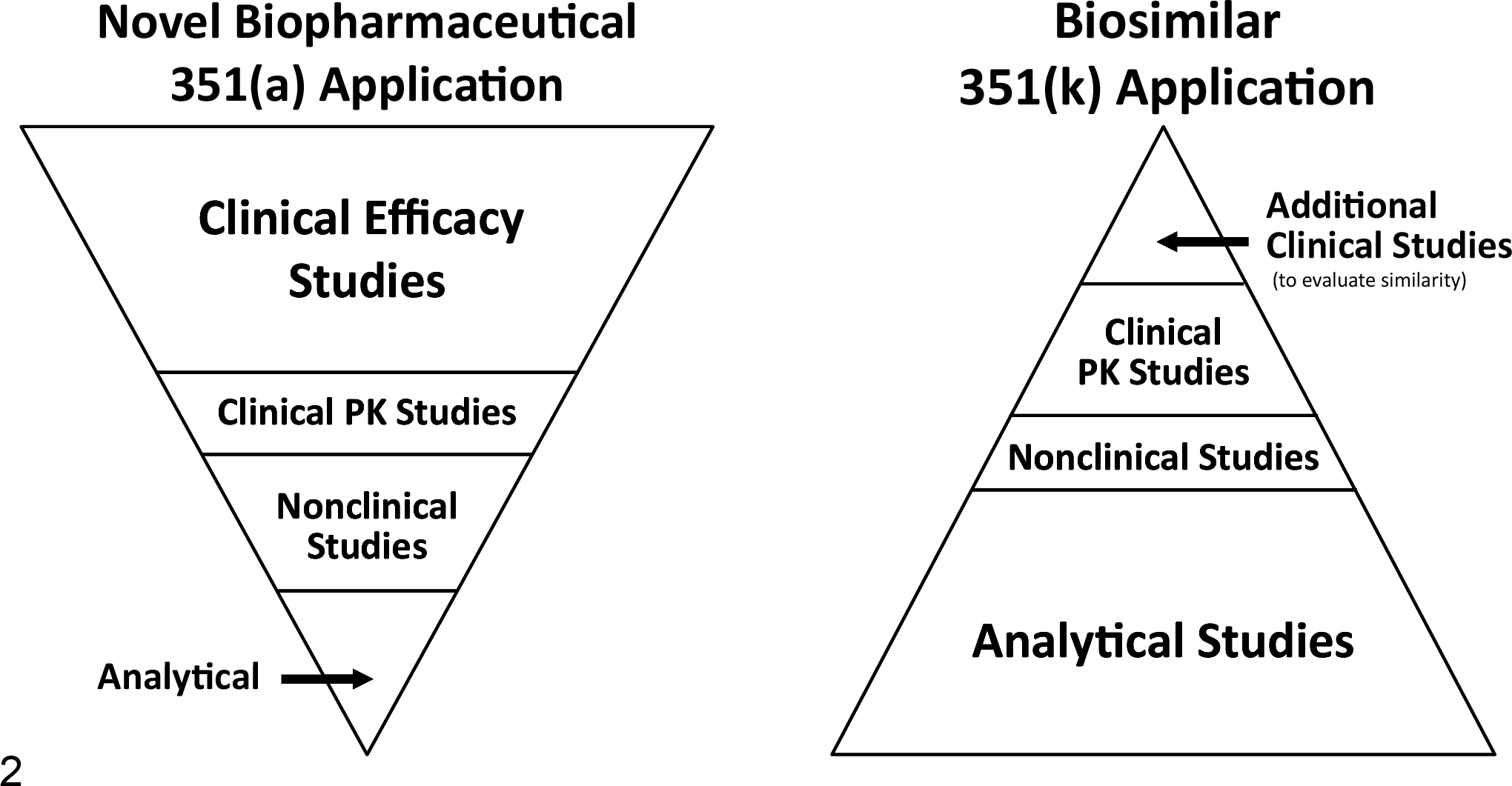
Relative comparison of the analytical, nonclinical, and clinical components in the license application for a novel biopharmaceutical and a biosimilar in the United States. The analytical characterization of a biosimilar is much more extensive than that conducted for a potential new biologic, while the nonclinical in vivo studies are reduced in number.
The nonclinical and clinical evaluations provide confidence that any differences observed at the quality level have no impact on the safety or efficacy of the biosimilar when compared to the reference product. The limited in vivo nonclinical studies (and greater reliance on in vitro product characterization) reflect the relative lack of sensitivity in detecting small differences between the reference product and biosimilar in in vivo toxicity studies under most circumstances. 32 In general, the clinical program will compare the efficacy, safety, pharmacokinetics, and immunogenicity of the biosimilar to the reference product, administered at the same dose and route of administration as the reference product. At least one clinical study, in the most sensitive patient population and measuring the most sensitive clinical endpoints, is required to identify any differences between the biosimilar and the reference product. 33 Regulatory guidelines are available to determine the extent of nonclinical and clinical data required, and the approval decision is made on a case-by-case basis, based on the totality of the data. 20 Additional detail on the quality and nonclinical considerations for biosimilar development are presented below.
Quality Considerations
The product characteristics and impurity profile of the innovator reference product are used as the target product profile for the biosimilar. The same formulation and cell line used in the reference product are not required; however, these differences make it more challenging to match product profiles between the reference product and the biosimilar.
Extensive comparability assessments are conducted, including confirmation of primary amino acid sequence, characterization of posttranslational modifications and higher-order (secondary and tertiary) structures, biophysical and biochemical characterization, and cell-based potency, binding kinetics, and functional assays. These assessments utilize state-of-the-art analytical instrumentation to characterize both the reference product and the biosimilar to ensure that any differences are well characterized and clinical relevance is well understood. 3
Different lots of the reference product, collected over time and representing a range of expiry dates, are examined to understand the interbatch variability to establish the appropriate ranges for the biosimilar specifications. Slight variation is possible among different commercial lots of the reference product, produced with different batches of medium or through different manufacturing sites. 3 In some cases, “manufacturing drift,” the result of manufacturing process changes over time, can be detected in lots of the reference product, as reported for rituximab and etanercept between 2007 and 2011. 30 The specific analytical tests are selected to maximize the potential for detecting any relevant differences in quality attributes, and typically orthogonal methods are used to characterize the same attribute; this maximizes the possibility that differences, if they exist, will be detected.
The analytical and functional assessments are iterative, assessing the biosimilar candidate throughout development and using the variation across contemporary commercial lots of the reference product to set the target bioprocess manufacturing specifications for the biosimilar. The extent of similarity between the reference product and the biosimilar also dictates the extent of clinical trials; despite extensive characterization, there is always some “residual risk” (small differences of uncertain significance and potential differences not detected with current methodology) that need to be further evaluated in clinical studies to demonstrate no effects on efficacy or safety. 20,34
Nonclinical in Vivo Considerations
In general, the nonclinical in vivo assessment of biosimilars is reduced, relative to novel biologics because similarity is typically demonstrated by more sensitive in vitro methods employed as part of the comparative quality characterization. Comparison of key nonclinical in vivo elements in the EMA and FDA biosimilar guidances pertaining to the development of mAb biosimilars is presented in Table 2.
Comparison of Key Nonclinical In Vivo Aspects of the EMA and FDA Biosimilar Guidances: Focusing on Monoclonal Antibodies.

Abbreviations: EMA, European Medicines Agency; FDA, Food and Drug Administration.
The small sample size in nonclinical animal studies, the inherent variability across animals, and the “normal” (vs “disease”) state of the animals make nonclinical in vivo studies relatively insensitive to detecting minor differences between the biosimilar and the reference product. The primary purpose of the nonclinical in vivo studies is to demonstrate that there are no biologically meaningful differences between the biosimilar and the reference product and that there are no new or unexpected toxicity findings. The nonclinical in vivo studies ensure safe progression into initial human studies.
Because the nonclinical studies are comparative in design per guidelines, they are larger than typical nonclinical safety studies for biologics. However, due to ethical considerations, these studies are usually not powered for statistical significance, and with mAb biosimilars, nonhuman primates are often the only relevant nonclinical species. Reduced study designs, incorporating 1 dose or 1 sex or excluding recovery, may be scientifically justified 22 but should be confirmed ahead of time with regulators. In some cases, noncomparative studies could be proposed to regulatory agencies; however, this requires a bridge to historical innovator data that may be either unavailable or outdated (eg, due to changes in immunogenicity assay formats), which may make it challenging to make valid comparisons to reference product data.
In addition to the larger study size, nonclinical biosimilar studies require 2 active molecules—the proposed biosimilar as well as the reference product—and there are costs and challenges in obtaining the reference product. Typically, the reference product must be sourced on the open market where one is competing with patients and other biosimilar developers looking to access sufficient material for the quality, nonclinical, and clinical programs (since the same comparator must be used throughout the development program). The availability of difference reference products in different geographies (eg, Rituxan in the United States and MabThera in the EU) also complicates the nonclinical study design; it may be possible to use EU-sourced material in the nonclinical in vivo studies if an analytical and clinical bridge across the products is established, but this should be confirmed up front with regulatory authorities.
The nonclinical in vivo program can be designed based on publically available information on the safety and efficacy of the approved reference product available in product labels, in regulatory documents (US Summary Basis of Approval, Canadian Product Monographs, and European Public Assessment Report), and in published research papers, nonclinical biopharmaceutical reviews, 1,7 patents, and meeting presentations and abstracts. In addition, available regulatory guidances (World Health Organization, Committee for Medicinal Products for Human Use, FDA) and scientific advice procedures in specific countries are helpful in designing the nonclinical program. In some cases, freedom-of-information requests to the innovator have produced partially redacted information on the reference product. Using the above information sources, a streamlined nonclinical program can be designed to replicate specific toxicity findings of the reference product and detect any clinically meaningful differences between the reference product and the proposed biosimilar.
Compared to the development of a novel biologic, nonclinical in vivo studies for a biosimilar are reduced in number as well as scope. Current guidances allow for situations where nonclinical in vivo studies are not necessary, 10,15 but most veterinary toxicologic pathologists feel that at least 1 in vivo study should be conducted prior to human dosing. If the bioanalytical and functional data from the quality exercise show no meaningful differences between the biosimilar and the reference product (or products), a single-dose pharmacokinetics study in a single sex may be sufficient. 22
Certain nonclinical studies, such as safety pharmacology, reproductive and developmental toxicity and carcinogenicity assessments, are not required by current guidelines.
Using available nonclinical safety information for the reference product, such as nonclinical species, study duration, and summary findings, comparative nonclinical safety studies can be designed to replicate the in vivo findings reported with the reference product. However, longer-duration studies (3 or 6 months) typically conducted for the reference product may not be needed for the proposed biosimilar if the nonclinical findings of the reference product are replicated in comparative studies of shorter duration (1 month). If no in vivo changes were reported in toxicity studies with the reference product, it may be possible to just evaluate comparative pharmacokinetics and immunogenicity in the nonclinical safety program. Whenever possible, pharmacokinetics and immunogenicity endpoints should be integrated with pharmacodynamic endpoints. In addition, nonclinical drug substance should be produced with the near-final manufacturing process to keep the number of nonclinical studies at a minimum.
In terms of nonclinical immunogenicity, the incidence of antibody formation is highly dependent on the sensitivity and specificity of the assay. 18,27 Additionally, the observed incidence of the antibody positivity in an assay may be influenced by several factors including assay methodology, sample handling, and timing of sample collection. Due to advances in assay methodology, it is better to do a contemporary comparative assessment of immunogenicity rather than rely on historical data available for the reference product. 18 As with all biopharmaceuticals, immunogenicity in animals is not predictive of immunogenicity in humans; 26 however, marked differences in the incidence of nonclinical ADAs between the reference product and the biosimilar may suggest that unrecognized differences between the 2 molecules exist.
Although biosimilars have been marketed for several years, information on the nonclinical studies to support these biologics is scant and typically focused on general principles related to analytical, toxicity, and immunogenicity assessments or utilizing blinded case studies. 2,24 More detailed descriptions have been described of nonclinical safety studies related to a biosimilar erythropoietin, conducted ahead of the EU regulatory framework but contributing to marketing approval in Eastern Europe. 25 More recently, nonclinical programs evaluating potential biosimilar anti-CD20 mAbs to rituximab have also been described. 12,28
Current and Future Challenges in Biosimilar Drug Development
While several biosimilars have been approved in the EU (including infliximab as the first mAb biosimilar), Canada, and Japan, the first biosimilar approval in the United States has not yet occurred. In addition, significant topics, such as extrapolation across disease indications, biosimilar naming, and interchangeability and automatic substitution, still need to be resolved across regulatory and health authorities.
Since a specific biopharmaceutical can be approved for more than 1 disease indication, extrapolation is the approval of a biosimilar for an indication for which it has not been evaluated in clinical trials. Extrapolation of clinical efficacy and safety data to other indications for the reference product that are not specifically studied during the clinical development of the biosimilar is possible based on the overall evidence of comparability provided by the 3-step comparability exercise, adequate scientific justification, and documented similar mechanisms of action. 23 Extrapolation is possible based on biosimilar guidelines 10,15 if there is a common mechanism of action across indications and if sufficient scientific justification is provided. In fact, the first mAb biosimilar approved in the EU (Remsima, an infliximab biosimilar) was evaluated in clinical trials of rheumatoid arthritis patients yet approved for all infliximab indications (Crohn disease, ulcerative colitis, ankylosing spondylitis, psoriatic arthritis, and arthritis), based on extrapolation across the various disease indications approved for the reference infliximab product. 13
Whether to use the same international nonproprietary name as the reference product or a unique identifier for a biosimilar is an area of active debate. Tracking adverse events associated with the use of reference versus biosimilar products will be difficult if the specific product or manufacturer cannot be easily identified; however, different names can undermine acceptance and utilization of biosimilars, reducing their overall uptake in society.
When 2 products can be exchanged for each other without significant risk of an adverse health outcome, the products are considered interchangeable. In the United States, the Biologics Price Competition and Innovation Act gives the FDA the ability to designate interchangeability for a biosimilar if specific changeover studies are conducted, 20 but this is not addressed in the EU biosimilar guidelines because it is outside the remit of the EMA. 10 Automatic substitution is the practice of substitution occurring at the dispensing level when the pharmacist is authorized to change a product without the prescribing physician’s prior consent. In the EU, decisions about automatic substitution are made by the individual countries, while in the United States, it is made at the individual state level. In both the EU and the United States, there is considerable difference of opinion about automatic substitution of biopharmaceuticals, in part because of a lack of information on potential immunogenicity issues associated with product switching.
Summary
Biosimilar development relies on a set of background data for the reference product that needs to be well understood to design an efficient development program. For global development, different guidelines and requirements may apply in different geographies, and navigating these guidelines requires up-front dialogue with regulators to clarify their expectations or negotiate an acceptable alternative. Comparative nonclinical in vivo studies are challenging due to the number of animals required and the limitations associated with obtaining the reference product (cost, quantity, procurement, and expiry). Despite these challenges, it is possible to customize the strategy for biosimilar development to the specific molecule, leveraging publically available information on the reference product and taking a weight-of-evidence approach in designing the nonclinical in vivo studies.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.