
Abstract
Chronic ingestion of yellow star thistle (Centaurea solstitialis) or Russian knapweed (Acroptilon repens) causes nigropallidal encephalomalacia (NPE) in horses with an abrupt onset of neurologic signs characterized by dystonia of lips and tongue, inability to prehend food, depression, and locomotor deficits. The objectives of this study were to reexamine the pathologic alterations of NPE and to conduct an immunohistochemistry study using antibodies to tyrosine hydroxylase and α-synuclein, to determine whether NPE brains show histopathologic features resembling those in human Parkinson disease. Results confirm that the NPE lesions are located within the substantia nigra pars reticulata, sparing the cell bodies of the dopaminergic neurons in the substantia nigra pars compacta, and in the rostral portion of the globus pallidus, with partial disruption of dopaminergic (tyrosine hydroxylase–positive) fibers passing through the globus pallidus. No abnormal cytoplasmic inclusions like the Lewy bodies of human Parkinson disease were seen in these NPE brains. These findings indicate that equine NPE may serve as a large animal model of environmentally acquired toxic parkinsonism, with clinical phenotype directly attributable to lesions in globus pallidus and substantia nigra pars reticulata rather than to the destruction of dopaminergic neurons.
Equine nigropallidal encephalomalacia (NPE) is a neurologic disease of horses that has been likened to human Parkinson disease (PD). 5,18 It was first described by Cordy in 1954 in California horses that were poisoned by ingesting yellow star thistle (Centaurea solstitialis). 5 Another poisonous plant, Russian knapweed (Acroptilon repens)—which grows in the western United States, Mediterranean countries, and Australia—has also been shown to cause the disease. 7,8,14,21,22 Typically, the clinical signs appear suddenly in horses, following prolonged ingestion of the poisonous plants, usually at least 28 to 35 days for A. repens and 33 to 81 days for C. solstitialis. 21,22 Invariably, paralysis of the lips and tongue is the first clinical presentation, incapacitating the horse’s ability to eat. There is reduced jaw tone, causing the mouth to remain partially open with the tongue protruding. Hypertonicity of facial and upper lip muscles, with exposure of upper teeth, is also a frequent observation. Other reported clinical signs include severe depression, with affected horses carrying their heads low. Ultimately, poisoned animals are unable to eat, become weak and emaciated, and eventually die of starvation. The histopathologic alterations of NPE have been well documented, consisting of circumscribed and mostly bilateral necrosis in the substantia nigra and the globus pallidus (GP). 5,7,8,14,21,22 However, there has been only limited ongoing research on NPE, and none has been able to identify unequivocally the proximate toxic principle in the 2 plants. 5,8,18,21 Repin, the most abundant constituent, is considered a leading suspect. 17
This clinical picture of affected horses has been likened to human PD in the veterinary literature. 5,18 The clinical defining features of human PD include resting tremor, rigidity, bradykinesia, and asymmetric onset, with symptom duration of at least 3 years, no atypical features, and a sustained response to levodopa or a dopamine agonist. 4 The neuropathologic criteria for diagnosis of PD are loss of dopamine neurons in the substantia nigra pars compacta (SNc) and Lewy body occurance. 6 The hypotheses of human PD pathogenesis have evolved over the years, and it is now believed that most PD cases are due to a combination of genetic and environmental factors that include exposures to drugs or toxic chemicals, 1 living in a rural environment, and consumption of well water. 4,6 Furthermore, several other human diseases, each with distinct neuropathologic features, exhibit parkinsonian symptoms (parkinsonism). These include progressive supranuclear palsy, multiple system atrophy, vascular parkinsonism, parkinsonism with dementia, corticobasal degeneration, and parkinsonism due to exposure to dopamine-blocking drugs (drug-induced parkinsonism). 4,6 In addition, neurodegenerative diseases with brain iron accumulation, also known as pantothenate kinase–associated neurodegeneration (PKAN) or Hallervorden-Spatz disease, 9,12 may present as parkinsonism and often include the characteristic Lewy bodies. 20
Given the complexity of the parkinsonian syndromes, it was the objective of this study to reexamine the neuropathologic alterations of NPE to determine whether this disease has similarities to any of the various human diseases that exhibit parkinsonism.
Materials and Methods
Case materials were either paraffin-embedded blocks or formalin-fixed brain from equine NPE cases from California and Colorado. Ten cases were examined (Table 1): 1 from the Colorado Animal Disease Laboratory and 9 from the California Animal Health and Food Safety Laboratory. Of these, 6 were male and 4 were female. All horses were older than 12 months. The Colorado case was due to A. repens intoxication while all California cases were caused by C. solstitialis intoxication. Initially, tissue blocks were sampled from as many brain regions as possible, including hippocampus, neocortex, cerebellum, thalamus, basal ganglia (including GP and striatum), midbrain (containing SNc and substantia nigra pars reticulata [SNr]), and pons (containing locus ceruleus). Control brain sections were taken from a 17-year-old mare in Michigan euthanized for complications associated with chronic cystitis and pyelonephritis.
Horses With Equine Nigropallidal Encephalomalacia a
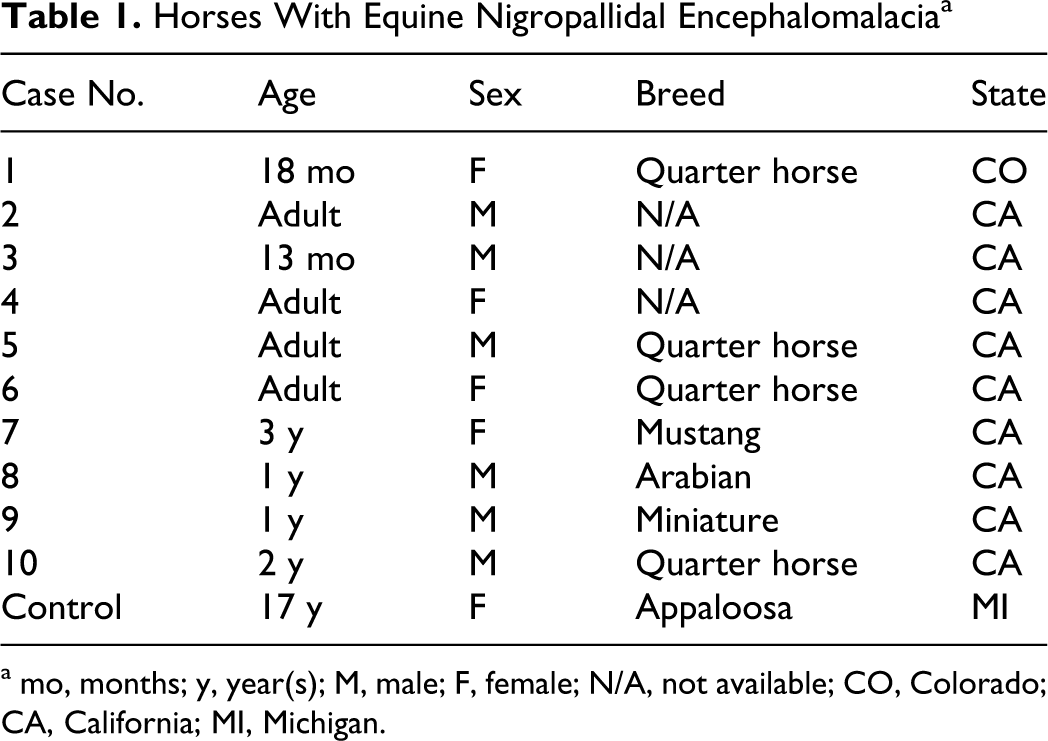
a mo, months; y, year(s); M, male; F, female; N/A, not available; CO, Colorado; CA, California; MI, Michigan.
Immunohistochemistry
Formalin-fixed, paraffin-embedded tissue sections (4 μm thick) were mounted on 2% 3-aminopropyltriethoxysilane-treated slides and dried at 56°C overnight. The slides were subsequently deparaffinized and rehydrated and underwent heat-induced epitope retrieval utilizing citrate buffer (pH 6.0) for 30 minutes at 100°C. The slides were rinsed in water and then immersed in 3% hydrogen peroxide / methanol bath for 30 minutes to block endogenous peroxidase. Following these pretreatments, the slides were subjected to standard avidin–biotin complex immunohistochemistry staining reactions performed at room temperature in a Dako autostainer, utilizing two 2-minute rinses between staining steps. The sections were incubated in the primary antibodies for 60 minutes, followed by appropriate biotinylated secondary antibodies for 30 minutes, the RTU Vectastain Elite ABC Reagent (Vector) for 30 minutes, and then developed using Nova Red (Vector) Peroxidase substrate kit for 15 minutes. The slides were rinsed in distilled water, counterstained with hematoxylin and eosin, rinsed, dehydrated through ascending grades of ethanol, cleared through xylene, and coverslipped using Flotex permanent mounting media. The primary antibodies used in this study were monoclonal mouse anti–tyrosine hydroxylase (at 10–30 μg/ml or 1:100 dilution of stock solution; Clone LNC1, Chemicon/Millipore, Temecula, CA) and polyclonal rabbit anti-alpha synuclein (at 1:1000 dilution of stock solution; Chemicon/Millipore). Positive labeling of SNc dopamine neurons in diseased and control horse brains (Fig. 1) served as internal positive controls for reactions for antibody to tyrosine hydroxylase (TH). Since α-synuclein is found normally in axon terminals, positive labeling of axon terminals in various brain regions in the horse brain sections, as well as Lewy bodies in human PD patients’ brains (data not shown), served as positive controls for antibody to α-synuclein.
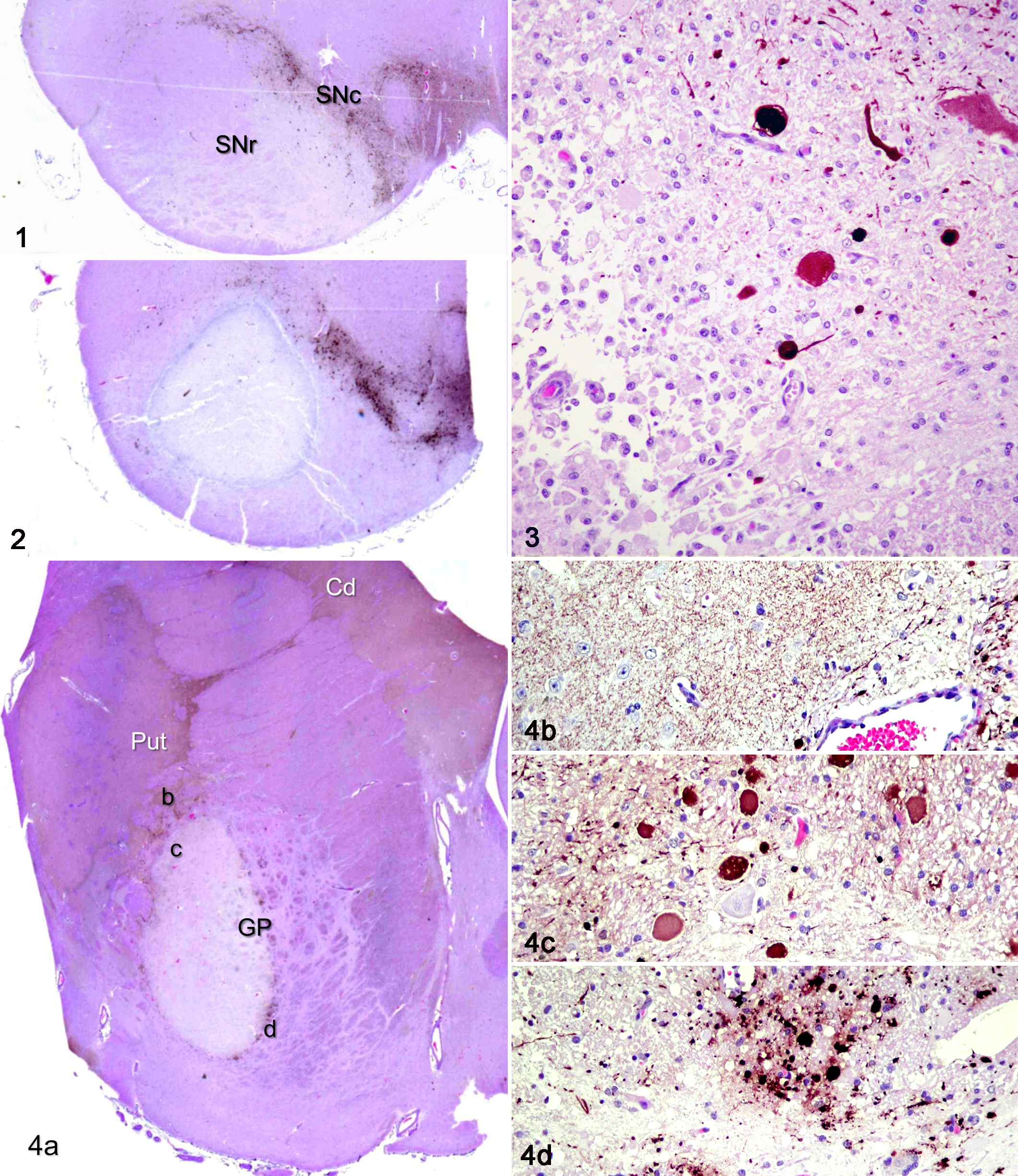
Midbrain; horse, control. The normal substantia nigra pars compacta (SNc) is outlined by tyrosine hydroxylase–positive (TH+) dopaminergic neurons and processes, in contrast to the substantia nigra pars reticulata (SNr). TH immunohistochemistry with HE counterstains.
Results
As reported in previous studies, 5,7,8,14,21,22 typical lesions of equine NPE consisted of well-circumscribed necrotic areas in the SNr (Fig. 2, 3) and GP (Fig. 4). Some lesions resembled subacute infarcts, rimmed or filled with macrophages (Fig. 3). A mild gliosis and many large eosinophilic spheroid profiles were seen along the rim of the lesions in both SNr and GP. Spheroids were also noted within some axon bundles adjacent to the lesion cores (Fig. 4c, 4d). A few lesion cores contained small vessels with thickened endothelium, as well as pale eosinophilic pyknotic neurons with very few macrophages in the neuropil, features suggestive of an early phase of an evolving ischemic infarct. Other lesions were more cystic, consistent with old infarcts. Many brains showed mineralized vessels within or near the necrotic lesions. There were no lesions noted in other areas of the brain.
Immunohistochemical staining with anti–tyrosine hydroxylase confirmed that the lesions were located within the SNr, with essentially no involvement of the dopaminergic neurons in the SNc (Figs. 1–3), and within the GP, with no involvement of the caudate or putamen (Fig. 4). The fine TH+ dopaminergic fibers in the striatum (caudate and putamen) appeared markedly reduced in some animals but relatively intact in others (Fig. 4b). However, there was a marked loss of en passant TH+ fibers within the core of the GP lesion that was at the same time rimmed with a number of enlarged neurites and spheroids, many (but not all) of which were TH+ (Fig. 4c, 4d).
Immunostaining for α-synuclein in sections from the midbrain and basal ganglia revealed a characteristic dense linear granular labeling pattern, with positive axon terminals outlining the negatively stained long dendrites of the GP 2,3 and SNr neurons (data not shown) in nonlesional areas, with scattered few positive-labeled profiles within the lesion cores. There was no evidence of any α-synuclein-positive inclusions that resembled Lewy bodies, hallmarks of human PD, 4,6 in any of the sections examined.
Discussion
Neuropathology of NPE Is Different From Typical Human PD
Our results confirmed that the NPE lesions are located within the SNr, sparing the cell bodies of the dopaminergic neurons in the SNc, and in the rostral portion of the GP, with partial disruption of dopaminergic (TH+) fibers passing through GP. These histopathology features are markedly different from typical human PD brains, in which the SNr and GP often appear relatively normal, whereas the number of pigmented dopaminergic neurons in SNc are variably but often markedly reduced in number. 6,16 The residual pigmented SNc dopaminergic neurons in PD brains often display characteristic cytoplasmic eosinophilic inclusions known as Lewy bodies that are immunoreactive for α-synuclein and ubiquitin. Cytoplasmic Lewy body–like inclusions were not evident in these equine NPE brains either with hematoxylin and eosin staining or α-synuclein immunostaining.
Similarity and Differences of Equine NPE to Other Human or Animal Neurologic Diseases
The various forms of neurodegenerative parkinsonism (eg, supranuclear palsy, multiple-system atrophy, parkinsonism with dementia, corticobasal degeneration) and parkinsonism due to exposure to dopamine blocking drugs are not associated with pallidal or nigral necrosis. The pathology of equine NPE most closely resembles the toxic injury to the GP that has been described in humans and experimental animals. Bilateral pallidal necrosis in humans is a well-known consequence of severe hypoxia or toxic injury due to carbon monoxide, manganese, methanol, or cyanide. 1,13 Similar nigral necrosis, often accompanied by pallidal necrosis, has been described with experimental seizures in rodents 10,11 and toxin ingestion in pigs. 15 Although the detailed mechanisms underlying these selective injuries have remained unclear, most theories have favored severe mitochondrial dysfunction in globus pallidal neurons. 1,20 The importance of mitochondrial function in GP neurons is further demonstrated by recent studies of the PKAN, a group of human autosomal recessive neurodegenerative diseases that preferentially affect bilateral GP. 9 The defective pantothenate kinase in PKAN results in deficiency of coenzyme A, which is critical for normal fatty acid metabolism in mitochondria.
Toxic Cause of Equine NPE
Previous studies have been unable to identify unequivocally the toxic principle in yellow star thistle and Russian knapweed. 5,8,18,21,22 Species differences in sensitivities to these plants have been demonstrated in that ruminants fed large quantities showed no ill effects, in contrast to horses. 17,21,22 Repin, the most abundant constituent isolated from these plants, is considered a leading suspect for causing equine NPE, 17 and it has been shown to reduce both cellular glutathione and mitochondrial functions in PC12 cells in tissue culture. 19 However, though toxic to rodents, repin has failed to produce distinctive pallidal lesions in laboratory rodents. 17 Further studies will be necessary to delineate the toxic mechanisms underlying equine NPE.
Conclusion
In summary, our findings confirm that equine NPE affects predominantly the GP and the SNr, with only partial disruption of nigrostriatal dopaminergic fibers passing through GP. The bilateral pallidal and nigral necrosis in NPE resembles the toxic injuries due to carbon monoxide, methanol, and cyanide in human. Our results also show that equine NPE does not cause abnormal cytoplasmic accumulation of α-synuclein, such as the Lewy bodies found in PD as well as those found in PKAN. 20 Thus, equine NPE may serve as a large animal model of an environmentally acquired toxic parkinsonism, with clinical phenotype directly attributable to lesions in GP and SNr rather than to the destruction of dopaminergic neurons as in the classic PD or other neurodegenerative parkinsonian diseases, such as progressive supranuclear palsy, multiple system atrophy, parkinsonism with dementia, or corticobasal degeneration.
Footnotes
Acknowledgements
A portion of this report was presented as an abstract at the 2008 annual meeting of the Society for Neuroscience, Washington, DC. Sabbatical release time to one of the authors (W.K.R.) from the Department of Pathobiology and Diagnostic Investigation is also appreciated. We wish also to thank the skillful assistance from the Michigan State University Investigative HistoPathology Laboratory.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported in part by an intramural research grant to HTC from the Department of Neurology and Ophthalmology of Michigan State University.