
Abstract
Weanling Brown Norway (BN) rats are susceptible to persistent steroid-responsive pulmonary abnormalities following resolution of an acute respiratory virus infection. In contrast, Fischer 344 (F344) rats recover without complications. Previous studies determined that NF-κB activation and subunit composition were markedly different between these 2 rat strains. This study examined whether viral infection also resulted in altered pulmonary expression of IκBα and IκBβ, 2 inhibitory regulators of NF-κB. Western blot analyses of total pulmonary protein extracted from BN and F344 rats at 7, 10, and 14 days after inoculation (n = 5 per group) did not reveal virus-induced differences in IκBβ expression. In contrast, a lower molecular weight form of IκBα appeared in the BN rats at 14 days postinfection, and it was still present at 21 days after infection (n = 5 per group). The change in IκBα expression observed in the susceptible BN but not the resistant F344 animals occurs when the epithelium is proliferating during the repair phase, and it correlates with the development of the persistent virus-induced airway inflammation and pulmonary functional abnormalities. These results further implicate differential regulation of NF-κB in the pathogenesis of virus-induced asthma.
Airway functional abnormalities, ranging from persistent increases in airway resistance and hyperresponsiveness to asthma, may develop following acute viral infections, especially in young children. 3,9 In particular, infections by respiratory syncytial and rhinoviruses have been associated with the development or exacerbation of asthma. 3,9 To better understand this outcome of virus infection, we investigated a well-described rat model of Sendai virus–induced persistent airway abnormalities. In both Brown Norway (BN) and Fischer 344 (F344) rat strains, Sendai virus infection induces acute necrotizing bronchiolitis and pneumonia. 5,6,10,12 F344 rats recover from infection without sequela. However, infection of BN rats results in long-term, chronic bronchiolar inflammation and fibrosis. 10 These structural abnormalities are associated with persistent asthma-like corticosteroid-responsive functional abnormalities, including elevated respiratory resistance, decreased dynamic compliance, and hyperresponsiveness to methacholine. 5,6,12
Cytokine-mediated recruitment and persistence of inflammatory cells has been investigated as a mechanism contributing to the development of the virus-induced asthma-like phenotype. Susceptible BN rats respond to viral infection with a Th2-like cytokine profile consisting of increased pulmonary expression of interleukin 4 (IL-4), IL-5, and IL-10, whereas the resistant F344 rats are characterized by a Th1-like phenotype in that viral infection induces a strong interferon-γ (IFN-γ) response. 1,11,13,15 –17 The importance of the IFN-γ response was confirmed by the finding that the development of bronchiolar fibrosis, persistent airway inflammation, and associated pulmonary functional abnormalities can be prevented by administering IFN-γ to BN rats before and during viral infection. 11 BN rats also respond to viral infection with a dramatic increase in pulmonary levels of tumor necrosis factor α (TNF-α) mRNA and protein not seen in the F344 strain. 17 Inhibition of TNF-α through use of a fusion protein containing the TNF receptor decreased the number of inflammatory cells and proliferating peribronchiolar fibroblasts in BN rats but not F344 rats, indicating differential expression of TNF-α as a major mediator of susceptibility to virus-induced airway abnormalities. 15,16
TNF-α and the transcription factor NF-κB are closely linked during the proinflammatory response to viral infection. 15,17 Previous studies revealed constitutive and virus-induced differences in pulmonary expression of NF-κB subunits between BN and F344 rats and implicated differential expression of NF-κB in the genetic susceptibility to persistent virus-induced airway abnormalities. 15 Activation of NF-κB is induced by viral proteins and inflammatory mediators, and its function is tightly regulated by the form and proteolysis of its associated inhibitor IκB. With this in mind, we performed studies to characterize pulmonary expression of 2 common NF-κB inhibitors: IκBα and IκBβ during the development of virus-induced chronic airway abnormalities.
Materials and Methods
Animals, Infection Protocol, and Experimental Design
Specific-pathogen-free 22-day-old weanling male BN and F344 rats were purchased from Harlan Sprague Dawley, Inc (Indianapolis, IN). They were negative for serum antibodies to Sendai virus, pneumonia virus of mice, mycoplasma, Kilham rat virus, sialodacryoadenitis virus (rat coronavirus), and respiratory aerobic bacteria. The rats were separated into experimental groups and individually housed in ventilated isolation units. After acclimation, the animals in the virus-infected groups were exposed to chorioallantoic fluid containing Sendai (parainfluenza type 1) virus at a concentration of 1 to 3 plaque-forming units per ml of gas for 15 minutes as previously described. 10 Uninfected rats were exposed to sterile chorioallantoic fluid. Virus-inoculated rats were processed at 7, 10, 14, and 21 days postinoculation (dpi) and sham-inoculated animals at 7, 14, and 21 dpi. Five animals were in each group, except at 14 dpi, when 10 BN rats were processed. In a separate infection experiment, 4 BN rats were injected intraperitoneally with 200 μg/g 5-bromo-2′-deoxyuridine (BrdU, Sigma Chemical Co, St Louis, MO; 2 mg/ml in phosphate buffered saline) 12 hours before being sacrificed at 10 dpi. All rats were deeply anesthetized with sodium pentobarbital and killed by exsanguination via intracardiac puncture. Lungs were removed and frozen in liquid nitrogen and stored at –80° C until protein was extracted for Western analysis.
Western Analysis
Lung tissue from the rats were homogenized in cold lysis buffer (4 ml/g tissue: PBS, 0.6% NP-40, 1 mmol EDTA, 150 mmol/liter NaCl, 1 mmol/liter EDTA, 10 mmol/liter HEPES [pH 7.9]); 0.02 ml of 0.1 M stock PMSF/gram of tissue was then added, and the whole cell lysate was harvested by centrifugation (10,000 rpm, 10 min, 4°C). For electrophoresis, 20 μg of the whole cell lysate was mixed with an equal volume of electrophoresis sample buffer (40% glycerol, 20% β-mercaptoethanol, 12% sodium dodecyl sulfate, 1.25 ml 250 mM Tris-HCl [pH 6.7], 0.05% bromophenol blue) and boiled for 90 seconds. Samples were fractionated on 10% polyacrylamide gels and transferred to nitrocellulose membranes with a transblot apparatus (14 V, overnight at 4°C; Biorad, Hercules, CA). Nonspecific binding was blocked by soaking the membranes in a blocking buffer solution (1×TBS, 0.05% Tween-20, 5% milk) for 15 minutes. The membranes were incubated (1 hour, room temperature) with either polyclonal rabbit anti-human IκBα or IκBβ (Santa Cruz Biotechnology, Inc, Santa Cruz, CA) at a concentration of 1 mg/ml in blocking buffer, washed 3 times in TBS with 0.05% Tween-20 (5 minutes, room temperature). Protein bands were detected with Chemiluminescence Luminol Reagent (Santa Cruz Biotechnology) according to the manufacturer’s instructions.
Two-Dimensional Electrophoretic Analysis
Two-dimensional analysis was performed as previously described on total pulmonary protein isolated from control (14 dpi) and infected BN rats (10, 14, 21 dpi) and F344 rats (14 dpi). 18 Protein samples were characterized from 5 animals per group. In brief, protein extracts (20 μg) were immunoprecipitated with the polyclonal IκBα antibodies, reduced with 1% β-mercaptoethanol, and denatured with 0.3% sodium dodecyl sulfate. Samples were loaded onto an isoelectrofocusing gel (pH, 4–8; Millipore, Billerica, MA) and run for 20,000 Vh. The second dimension was performed as previously described on a 10% acrylamide gel. 18 Relative isoelectric points were determined by parallel migration of carbamylated muscle creatine phosphokinase standard (BDH, West Chester, PA), and the molecular weights of the detected proteins were determined by comparison with standardized markers. After sodium dodecyl sulfate polyacrylamide gel electrophoresis, proteins were blotted as described for one-dimensional polyacrylamide gels.
BrdU Detection
The rats were injected with BrDU on day 9.5 and the lungs were harvested, perfused and fixed with 4% paraformaldehyde as previously described. 17 They were embedded in paraffin within 12 hours. Four transverse 5-μm-thick sections were cut from each lung, mounted on slides, deparaffinized, and immunocytochemically stained for BrdU with a mouse monoclonal anti-BrdU antibody (Becton-Dickinson, Mountain View, CA) at 1:100 for 1 hour as previously described. 17
Results
To characterize IκBα and IκBβ, Western blot analyses were performed on whole pulmonary protein extracts harvested from Sendai virus–inoculated BN and F344 rats at 7, 10, 14, and 21 dpi and from sham-inoculated animals at 7, 14, and 21 dpi. Five animals per group were evaluated at each time point, with the exception of 14 dpi, when IκBα expression was assessed in an additional 5 virus-inoculated BN rats. No qualitative differences in IκBβ expression were detected in any of the animals, regardless of strain (Fig. 1A). In contrast, Western blot analyses of pulmonary proteins isolated from BN rats revealed that between 10 and 14 days after virus inoculation, the predominant IκBα band shifted to a form with slightly lower molecular weight (Fig. 1B). This form of IκBα was present in some of the BN rats as early as 10 dpi and was still present at 21 dpi (Figs. 1C). It was faintly detected in the BN rats at other time points after viral infection and occasionally in the uninfected BN rats (Fig. 1C); however, it was not observed in the virus- or sham-inoculated F344 rats. In addition, two-dimensional gel electrophoresis of pulmonary protein extracted from infected BN rats identified a new form of IκBα at 14 and 21 dpi and occasionally at 10 dpi (Fig. 2A, 2B).
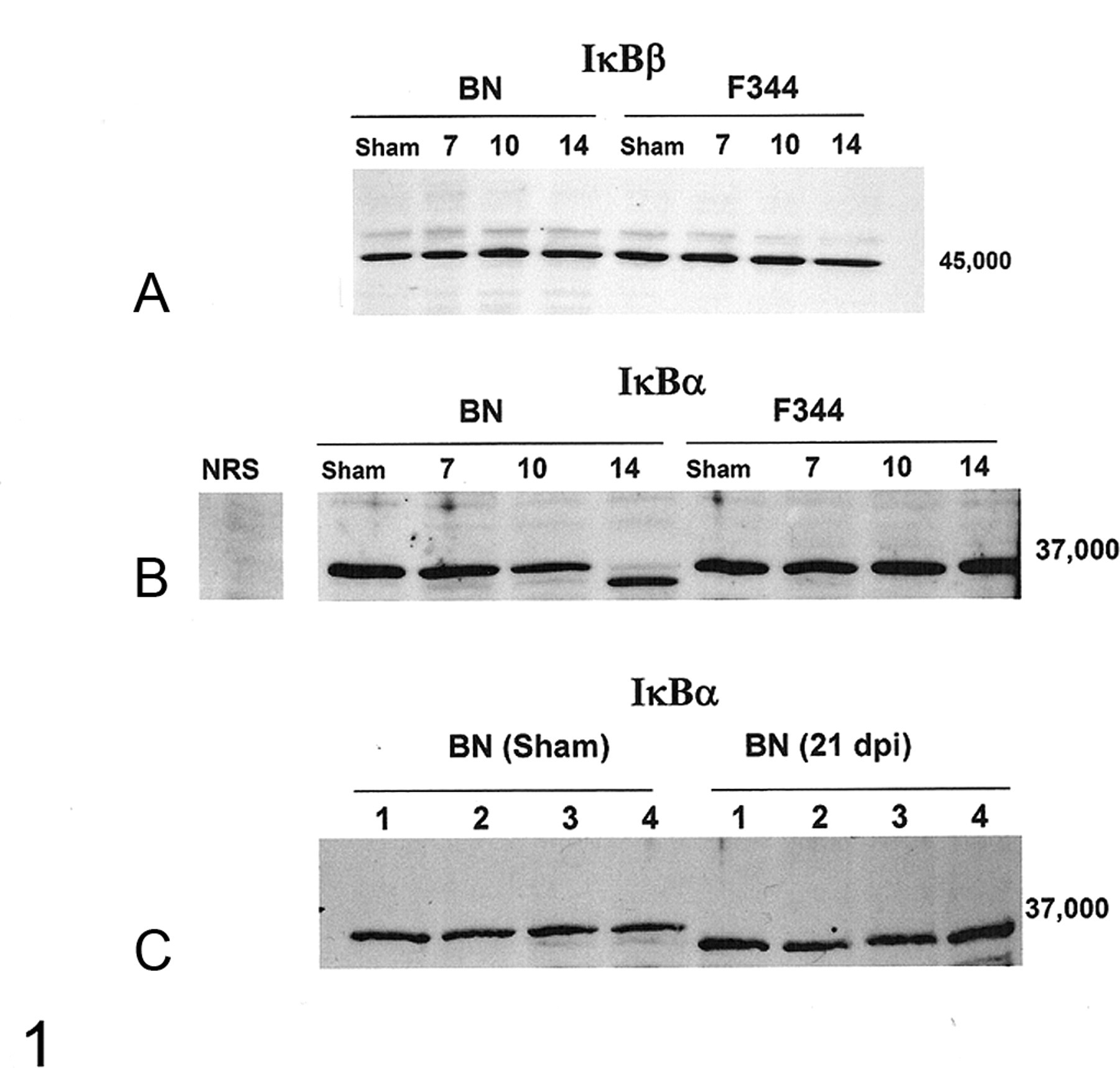
Western analysis for IκBα and IκBβ performed on total protein isolated from lung tissue reveals no virus-induced changes in expression of IκBβ (A) and that all Brown Norway (BN) rats had a shift in the predominant form of IκBα 14 days after virus inoculation (B). Western analysis was performed on protein extracted from 5 animals per group for all time points except 14 days after virus inoculation, when expression was analyzed for IκBα in 10 BN rats (representative gel shown, B). C, the lower molecular weight form was still the predominate pulmonary form of IκBα in the BN rats (n = 4) 21 days after viral infection; see the results from 4 sham animals and note that all the IκBα bands have shifted to the lower molecular weight form in the infected animals. Separate gels are shown (A–C), and the results represent a series of 5 gels, each containing samples from different animals. All samples shown together were run on the same gel. Fischer 344, F344; NRS, Normal Rabbit Sera; dpi, days postinoculation.
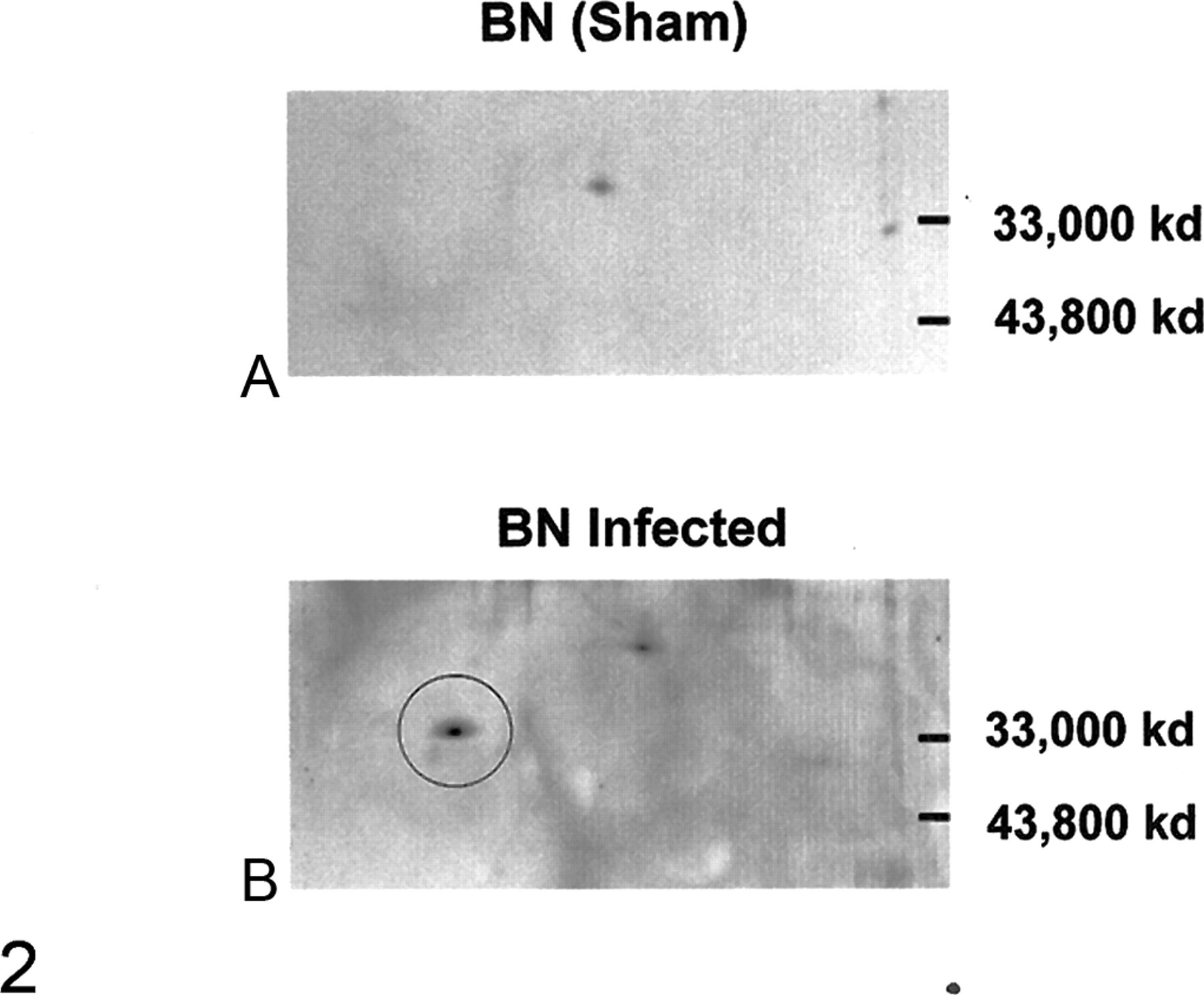
Two-dimensional gels with antibodies against IκBα were performed on total pulmonary protein isolated from control Brown Norway (BN) rats (14 days postinoculation [dpi]), infected BN rats (10, 14, 21 dpi), and Fischer 344 rats (14 dpi). Protein samples from 5 animals per group were characterized. An extra protein density (circle) was identified in samples from infected BN rats at 10, 14, and 21 dpi but was not in any of the samples from F344 rats or control BN rats. Representative gels from control (A) and virus-inoculated (B) BN rats are shown, 14 dpi.
The change in the predominant pulmonary form of IκBα occurred between 10 and 14 dpi, after the peak of the acute inflammatory response at 7 dpi. The shift in the pulmonary form of IκBα was complete and persistent, which implies that it was occurring in a large population of resident cells as part of the repair response. In addition, double bands were noted in some of the BN rats at 10 dpi (Fig. 3 ), indicating that the change was likely occurring closer to 10 dpi than 14 dpi. To characterize changes in the resident pulmonary cell populations during the transition in IκBα expression, BrdU labeling was performed to identify proliferating cell populations. The most pronounced labeling was observed in the airway epithelial cells (Fig. 3).
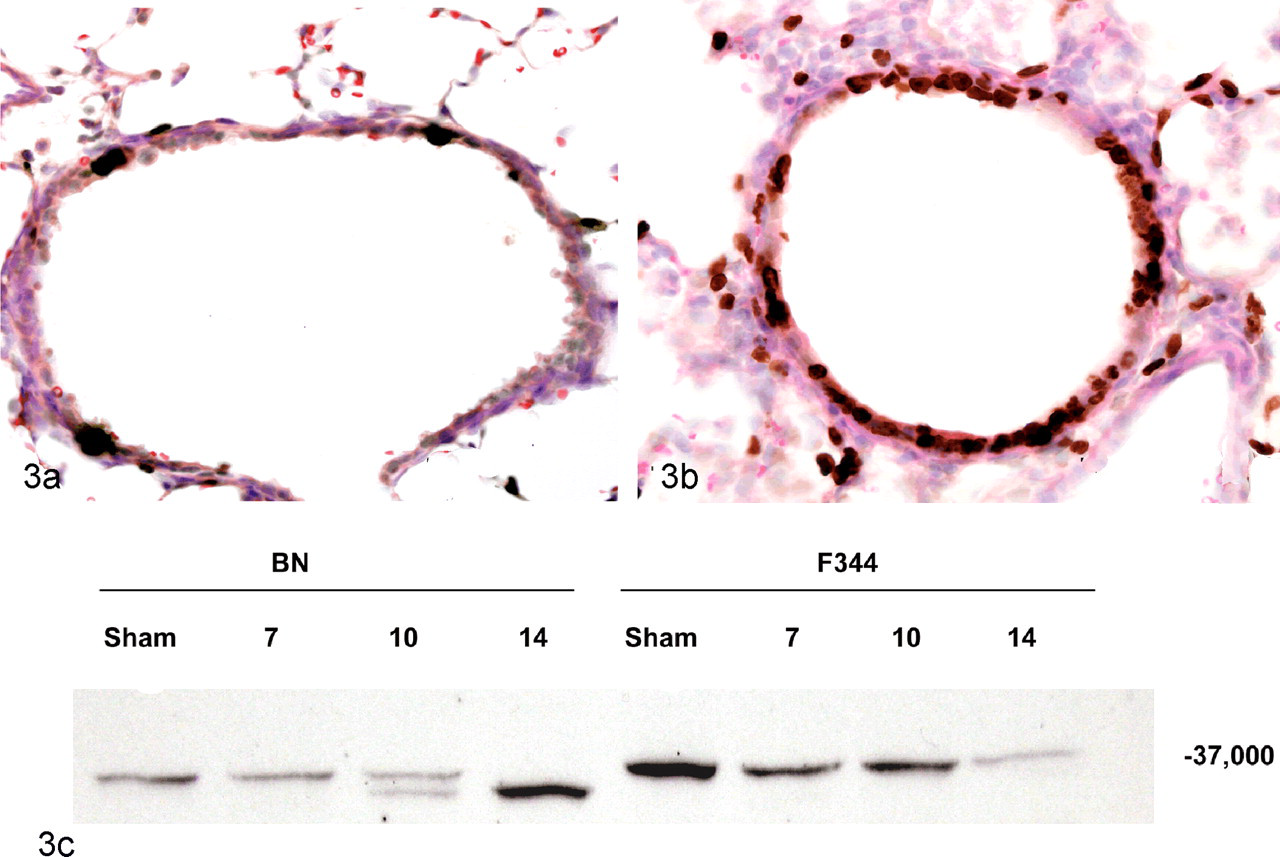
Lung tissue from Brown Norway (BN) rats 10 days after sham (A) or Sendai virus (B) inoculation. Viral infection induces proliferation of bronchiolar airway epithelial cells, as indicated by the marked increase in BrdU labeling. The airway epithelial cell proliferation corresponds to when the predominant pulmonary form of IκBα is transitioning to a lower molecular weight form, as observed in the Western gel (C). Fischer 344, F344. Immunohistochemical labeling of BrdU with immunoperoxidase detection and HE counterstain.
Discussion
Differential NF-κB activation and subunit composition in the lungs have been linked to susceptibility or resistance to chronic postviral airway abnormalities. 15 There are several different forms of NF-κB, which can be present as heterodimeric and homodimeric molecules, the most common of which contains the p65 and p50 subunits. 2 F344 rats, which do not develop the asthma-like phenotype following Sendai virus infection, predominantly express the NF-κB p65 subunit in the lungs, and virus infection temporarily increases expression of the p50 subunit. 15 In contrast, the susceptible BN rats predominantly express the p50 subunit and have higher levels of virus-induced NF-κB activation than do the F344 rats both preinfection and postinfection. 15 In addition, the development of postviral airway abnormalities can be prevented in the BN strain by 2 treatments that result in increased pulmonary expression of the p65 subunit. 15 These findings implicate the expression and composition of NF-κB in the susceptibility to and pathogenesis of virus-induced persistent airway disease. To further characterize the link, this study examined the expression of 2 important NF-κB regulators: IκBα and IκBβ. We found that in the BN rats, which are susceptible to virus-induced asthma-like phenotype, the predominant pulmonary form of IκBα changed to a slightly lower molecular weight form between 10 and 14 dpi (Figs. 1, 3), whereas expression of IκBβ was similar across all the groups.
BN rats have an allergic phenotype, and Sendai virus infection increases pulmonary expression of predominantly Th2 immune mediators, including IL-4, IL-5, IL-10, TNF-α, and TGF-β1. 16,17 In contrast, F344 rats are Th1 (IL-12, IP-10, IFN-γ) responders. 1,11,13 The greatest increase in these inflammatory mediators occurs during the peak of the acute inflammatory response, around 7 dpi, and all return to, or are close to, baseline expression levels by 14 dpi. Total pulmonary activation of NF-κB has a similar profile and peaks at 7 dpi. 15 The differences in pulmonary NF-κB expression between BN and F344 rats at 14 dpi are interesting: At this time, the majority of the NF-κB binding DNA in BN rats is due to forms containing the p50 subunit, whereas the p65 subunit is predominant in the F344 rats. 15 How these differences in NF-κB subunit predominance are related to the shift in IκBα are unclear because IκBα associates with forms of NF-κB containing both p65 and p50. 2 Regardless, the virus-induced change in IκBα is the only documented change in an inflammatory regulator that occurs during the resolution of the acute virus-induced inflammatory response and that persists after resolution of the infection.
Because the lower molecular weight form of IκBα does not occur in the F344 rats, was occasionally detected in the uninfected BN rats and persists in the infected BN rats, the change in IκBα is most likely a triggered host response, rather than an effect that requires the actual presence of the virus. The acute inflammatory response to Sendai virus infection resolves between 10 and 14 dpi in both rat strains. 10 Although the overall inflammatory cellular infiltrate is markedly reduced, at 14 dpi BN rats have 3-fold greater numbers of bronchioles with aggregates of lymphocytes and macrophages and increased numbers of bronchiolar mast cells than do F344 rats. 10 The airway inflammation persists in the BN rats and is associated with steroid-responsive pulmonary functional abnormalities. 5,6 It is possible that the change in pulmonary IκBα expression is directly associated with the cells making up the persistent inflammatory infiltrate; however, this cell population is multifocal, and a more likely explanation for such a complete shift in expression is that it is occurring in a common resident cell population. Two possible cell populations are airway epithelial cells and macrophages. There were no significant differences in the total number of pulmonary macrophages between infected and control BN rats at 14 dpi, implying that the change in IκBα is not associated with increased numbers of macrophages. 10 Although not definitive, BrdU labeling implicates epithelial cells because there is marked proliferation of airway epithelial cells during the period when pulmonary form of IκBα is transitioning to a lower molecular weight form (Fig. 3). Unfortunately, IκBα is expressed by most cells, and immunohistochemistry cannot be used to distinguish the 2 different molecular weight forms.
Several attempts to sequence the altered form of IκBα from the two-dimensional gels were unsuccessful, which suggests that posttranslational modifications may have blocked the N terminus. Two common posttranslational modifications of IκBα that can produce doublet bands are phosphorylation and oxidation. Phosphorylation leads to immediate degradation of IκBα and activation of NF-κB. 8 Phosphorylated IκBα is therefore transient and unlikely to be present as a persistent change. In contrast, oxidized IκBα can persist; it runs slightly higher on Western gels than does the unoxidized form, producing a doublet similar to that observed in the infected BN rats. 4,7,14 Oxidation of IκBα occurs on Met45, which prevents degradation by blocking phosphorylation of Ser32 and Ser36, thus inhibiting activation of NF-κB. It is caused by a variety of agents, including several neutrophilic and eosinophilic enzymes, and it is thought to be a mechanism for dampening NF-κB-induced inflammation. 4,7,14 Oxidized IκBα can be reduced by methionine sulphoxide reductase, a common and important enzyme controlling cellular oxidative stress. 7 Of interest in the rat model, especially given the importance of TNF-α in the development of the asthma-like phenotype, 17 is the finding that oxidation of IκBα inhibits TNF-α-induced NF-κB activation. 14 A change to an unoxidized form of IκBα could therefore enhance activation of NF-κB and contribute to the persistent airway inflammation in the BN rats.
Given our results in view of both the traditional NF-κB activation pathway and the recently recognized importance of NF-κB and IκBα in epigenetic regulation of gene expression, a persistent virus-induced change in the predominate form of IκBα could result in easier localized activation of NF-κB within the airways and thus contribute to the persistent airway abnormalities through 2 different mechanisms: The first is that the change in IκBα lowers its phosphorylation threshold, making NF-κB more easily activated; the second is that it enhances NF-κB gene transcription by binding to histone deacetylase 1 and 3 and inactivating them by transporting them out of the nucleus, thus increasing the time that NF-κB and, potentially, other transcription factors are bound to promoter sequences (Fig. 4 ). 19
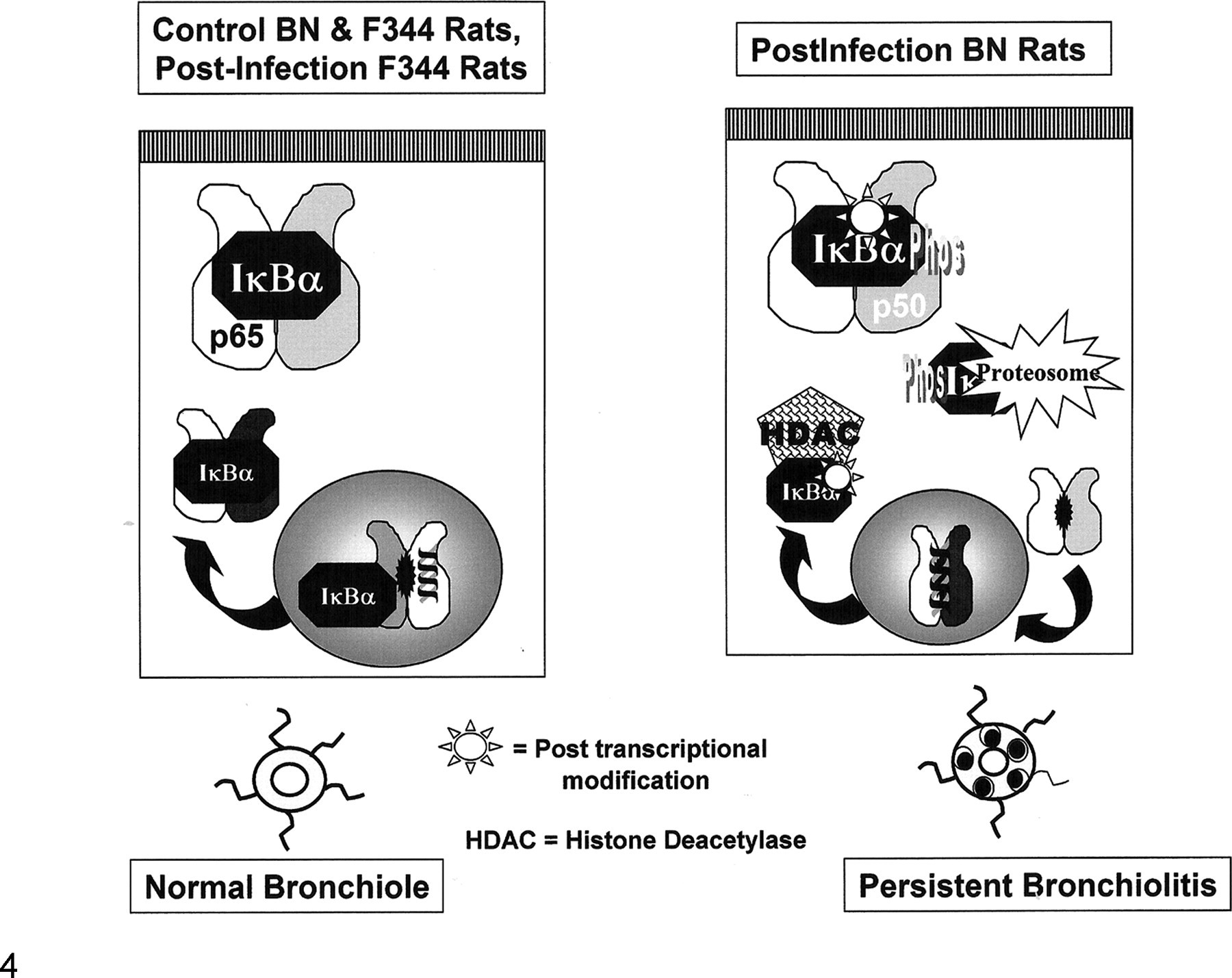
Schematic of the potential effects of the persistent virus-induced modification of IκBα. The modification is most likely posttranscriptional (possibly, the loss of an oxygen group on Met 45), occurring in the infected Brown Norway (BN) rat during the period of transition between the acute inflammatory response to the virus and the establishment of the chronic persistent airway inflammation and functional abnormalities. The shift in IκBα expression is complete, and it corresponds to when the epithelium is proliferating during the repair response. IκBα can affect gene expression through 2 mechanisms: One is through its phosphorylation and degradation, which allows NF-κB to move into the nucleus and bind DNA; the other is by increasing the time transcription factors that are bound to promoter sites by removing histone deacetylase 1 and 3 from the nucleus, thus prolonging gene transcription. Virus-induced predominance of a form of IκBα in the lung that either facilitates a lower threshold for NF-κB activation or prolongs gene transcription could be contributing to the persistent airway inflammation characteristic of the asthma-like phenotype that develops in the BN rats postinfection. Fischer 344, F344.
The results of the present study indicate that genetic and virus-induced differences in IκBα are associated with susceptibility to persistent virus-induced airway abnormalities. The results also support those of other studies implicating differential expression and activation of NF-κB in the development of chronic postviral airway disease. 18 Finally, they indicate that a virus-induced change in the epigenomic regulation may be occurring, potentially in epithelial cells.
Footnotes
The authors declared that they had no conflicts of interest with respect to their authorship or the publication of this article.
This project was funded by a University of Florida College of Veterinary Medicine Research Development Award and University of Georgia faculty start-up funds.