
Abstract
Anaplastic Lymphoma Kinase (ALK) is a potent oncogenic driver of lung adenocarcinoma (LUAD). ALK is constitutively activated by gene fusion events between the ALK and other gene fusion partners in about 2-3% of LUADs, characterized by few other gene alterations. ALK-fusions are a druggable target through potent pharmacological inhibitors of tyrosine kinase activity. Thus, several ALK-TKIs (Tyrosine Kinase Inhibitors) of first-, second- and third-generation have been developed that improved the outcomes of ALK-rearranged LUADs when used as first- or second-line agents. However, resistance mechanisms greatly limit the durability of the therapeutic effects elicited by these TKIs. The molecular mechanisms responsible for these resistance mechanisms have been in part elucidated, but overcoming acquired resistance to ALK-derived therapy remains a great challenge. Some new therapeutic strategies under investigation aim to induce long-term remission in ALK-fusion positive LUADs.
Introduction
Lung cancer is the main cause of cancer mortality worldwide. Non-small cell lung cancer (NSCLC) accounts for about 85% of lung cancers; two main histological types of NSCLC have been described: lung adenocarcinoma (LUAD) (40-50% of lung cancers) and lung squamous cell carcinoma (LUSC) (25-30% of lung cancers).
The development of next generation sequencing techniques has led to the identification of the most recurrent genetic alterations observed in LUAD, allowing the identification of biomarkers required for the selection of LUAD patients for chemotherapy, targeted therapy or immunotherapy. LUAD is among the cancers bearing the highest number of different mutated genes, with a high tumor burden.1-2 LUAD is associated with smoking-related mutational signatures 2 and 5. 3 Finally, LUAD is a tumor with moderate genetic differences between primary and metastatic state. 4
TCGA (The Cancer Genome Atlas) reported an extensive characterization of genetic alterations observed in LUAD, showing that these tumors have a high mutation burden (8.8 mutations/megabase) and have a large set of recurrently mutated genes: frequently mutated were TP5, KRAS, EGFR, STK11, KEAP1, NF1, BRAF, SETD2, RBM10, MGA, MET, ARID1A, SMARCA4, RB1, CDKN2A, U2AF1. 5 The analysis of mRNA sequencing showed the presence of aberrant mRNA transcripts due to gene fusion events involving ALK, ROS1 and RET genes (each occurring in about 1-3% of cases). 5
The mutation profile is different in smoker and non-smoker LUAD patients. The average mutation frequency was more than 10-fold higher in smokers than never-smokers. 6 Mutations in EGFR, ROS1 and ALK preferentially occur in non-smoking patients, whereas mutations in TP53, KRAS, BRAF, BRAF, JAK2 and JAK3 mainly occur in smokers. 6
This article will review recent developments in the analysis of the biological characteristics of ALK fusion positive LUADs and the progresses in the development of new therapeutic approaches of these tumors based on the generation of potent TKIs. This review will also highlight the molecular heterogeneities of these tumors that have to be carefully considered for a prognostic evaluation and for the definition of an optimal individual strategy of treatment.
ALK-rearranged LUAD
ALK (anaplastic lymphoma kinase) gene rearrangements are observed in about 2-8% of LUAD. ALK gene encodes a receptor transmembrane protein with tyrosine kinase activity. ALK gene is localized on chromosome 2 and makes part of insulin receptor superfamily. ALK protein is composed by three domains, extracellular, transmembrane and intracellular, with the intracellular domain mediating tyrosine kinase activity. The extracellular domain of ALK contains two MAM subdomains, a glycine-rich region and a LDLa subdomain. The physiological ligands of ALK are unknown; three putative ligands are represented by pleiotropin, midkine and augmentor. The binding of ligands to ALK induces receptor dimerization and autophosphorylation and activation of downstream signaling pathways, including PI3K-AKT, mTOR, MAPK and STAT3, resulting in induction of cell proliferation, survival and differentiation. 7
Three types of ALK gene alterations have been observed in human tumors: ALK gene rearrangements, ALK gene point mutations and ALK gene amplifications. In LUAD, the most common alteration consists in a translocation of ALK gene with another partner gene, determining a gene fusion event between the kinase domain of ALK and the amino terminal segment of different protein partners; the resulting fusion protein possesses an aberrantly constitutive kinase activity with receptor activation independent of ligand binding. In LUAD the most frequent ALK gene rearrangements involves a breakpoint at the level of exon 20 of ALK gene and the generation of a EML4-ALK fusion gene. 8
The EML4 gene encodes a protein belonging to the family of echinoderm microtubule-associated protein (EMAP)-like proteins and is composed by three different regions: A N-terminal basic region, a hydrophobic EMAP-like protein domain and a tryptophan-aspartic (WD) repeat domain.8-9 EML4 protein plays an important role in the generation and maintenance of the cellular microtubular network and in the control of cell survival and proliferation.8-9
Different breakpoints at the level of the EML4 gene generates different variants of the EML4-ALK fusions: the variant 1 involves the fusion of exon 13 of the EML4 gene with exon 20 of ALK gene; the variant 2 implies the fusion of exon 20 of ELM4 with exon 20 of ALK; the variant 3 involves the fusion of exon 6a or 6b of ELM4 with exon 20 of ALK (Figure 1). 10 In a study on 35 ALK-mutant LUAD patients, 54% displayed ELM4-ALK variant 1, 14% variant 2, 12% variant 3a/3b and 20% other rarer variants; in patients treated with the ALK inhibitor crizotinib, patients with variant 1 displayed a better PFS (progression-free survival) compared to those with other variants. 10 Lin et al. have explored 129 LUAD patients with ALK-positive tumors and showed that 43% of these patients have a variant 1 EML4-ALK, 2% variant 2, 40% variant 3a/3b, 2% variant 5a/5b, 4% variant 5’ and 1% variant 7: furthermore, 5% of patients have a non-EML4-ALK fusion. 11 This study failed to confirm that PFS time was significantly longer in patients with variant 1 than in those with non-variant 1; however, after first-line treatment with crizotinib, variant 1 patients responded better to second-line treatment with lorlatinib. 11
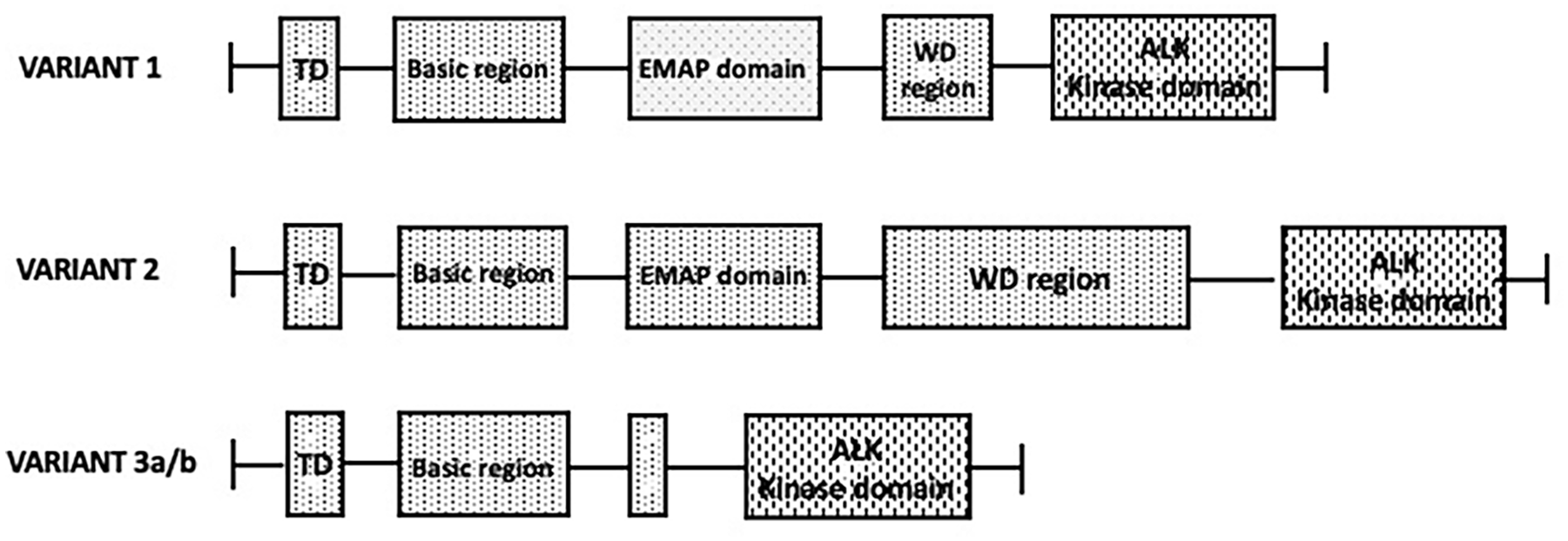
Molecular structure of the three main variants (1, 2 and 3a/3b) generated through the fusion of EML4 with ALK gene; structural domains of the EML4 and ALK genes are shown.
Desai et al. reported a detailed analysis of the genetic alterations observed in LUAD, as based on the screening of 10,082 patients; ALK gene alterations were observed in 5% of samples: 60% missense mutations; 45% fusions; 9% truncating mutations; however, functionally significant ALK gene alterations were observed in 2% of LUAD: 259 gene fusions and 25 missense mutations. 14 ALK fusions with EML4-ALK corresponded to 81.5% of all fusions. 12 Other studies have shown the occurrence of missense ALK point mutations in treatment-naïve NSCLC patients; thus, Ding et al. reported four ALK point mutations (P496L, P542R, S631I and V1135E) in 188 LUAD patients evaluated by targeted DNA sequencing. 13
Few studies have provided an integrated molecular characterization of large cohorts of these patients. A molecular characterization of 158 ALK-rearranged NSCLCs (95% LUADs) provided evidence that most of these tumors bear EML4-ALK gene fusions (31% variant 1, 10% variant 2, 31% variant 3). 14 EML4-ALK short isoforms (3 and 5a) were associated with more advanced stage and more metastases and with a trend for shorter PFS than long isoforms (1 and 2). 14 Analysis of the mutational profile showed that ALK-rearranged LUADs do not have concurrent mutations in EGFR, KRAS or rearranged RET or ROS1; the most frequent concurrent mutations in these tumors were TP53 (11%), ARID1A (7%), ATRX (6%), NF1 (6%). 14
Couetoux du Tertre et al. reported the comprehensive genomic landscape of 11 ALK-rearranged LUAD undergoing targeted therapy with crizotinib; TP53 was the gene most frequently co-mutated in these patients and was associated with short PFS. 15
Several studies have shown that TP53 mutations predict for poor survival in ALK-rearranged LUADs.16,17
Although driver gene mutations were reported to be mutually exclusive in LUADs, however, some driver genes seem to occur in association with EGFR mutations. Yang et al. explored in 977 NSCLC patients the presence of concomitant EGFR mutations and ALK rearrangements: concomitant EGFR and ALK alterations were observed in 13% of all patients, 3.9% of EGFR-mutant and 18.6% of ALK-rearranged patients. 18 Immunohistochemistry analysis showed the co-expression of mutant EGFR and ALK fusion protein in the same tumor cell populations. 18
Concomitant EML4-ALKfusion gene and KRAS mutations are observed in a minority of patients. Li and coworkers reported in 132 NSCLC patients with ALK rearrangement a frequency of 0.76% of KRAS mutations and 2.3% of EGFR mutations. 19
The fundamental role of EML4-ALK fusion gene in the pathogenesis of ALK-rearranged LUADs was directly supported through the study of transgenic mouse lines that express EML4-ALK specifically in lung alveolar epithelial cells: all these transgenic mice developed adenocarcinoma nodules in lungs a few weeks after birth. 20 These tumors were exquisitely sensitive to ALK-TKIs. 20 A transgenic conditional mouse model recapitulating ELM4-ALK-rearranged human NSCLCs provided a tool to explore the mechanisms of resistance to crizotinib, occurring after an initial response and involving mainly the acquisition of ALK G1202R mutation. 21
Methods for detection of ALK fusions
Five different methods can be used for the detection of ALK fusions in LUADs: immunohistochemistry (IHC), fluorescence in situ hybridization (FISH), RT-PCR (reverse transcriptase-polymerase chain reaction), DNA sequencing NGS (next generation sequencing) and RNA sequencing NGS. 22 IHC is based on the use of specific antibodies that interact with high affinity and specificity with ALK (ALK protein expression in undetectable or very low in normal lung tissue but is moderately or high in ALK-rearranged LUADs); the advantage of IHC is to detect ALK fusion expression and the limit is that the LAK fusion partner remains unknown; IHC may give rise to false positivity in some instances, such as in LUADs with rich mucin component. 23 FISH is a specific technique of moderate sensitivity and of high specificity for detecting ALK rearrangements; the limits of this technique are that expression cannot be evaluated and the ALK fusion partner cannot be identified. RT-PCR is a highly specific and sensitive technique for ALK fusion detection; however, this technique may generate false negative cases due to the detection of only known fusion partners; this technique detects expression of the fusion ALK gene. DNA-based NGS is a specific and sensitive technique for detection of ALK fusion; the advantages of this technique are related to the capacity to detect multiple genetic alterations in a tumor DNA sample and to detect both known and unknown ALK fusion partners. RNA-based NGS, either amplicon-based NGS or whole transcriptome NGS allows the detection of ALK fusions expression at RNA level with high sensitivity and specificity and can detect unknown ALK fusion partners.
Target therapy of ALK-rearranged LUADs
The central and key role played by ALK rearrangements, and the substantial absence or limitation of other driver gene mutations strongly have supported the rationale of treating ALK-rearranged LUADs with agents targeting ALK fusion proteins. Four generations of ALK tyrosine kinase inhibitors have been developed and clinically evaluated: first generation (crizotinib); second generation (ceritinib, alectinib, brigatinib and ensartinib); third generation (lorlatinib); fourth generation (TPX-0131 and NVL-655).
Crizotinib
Crizotinib was the first ALK TKI introduced in the therapy of ALK-rearranged LUAD. Two large phase III clinical trials have documented the superiority of crizotinib compared to standard first-line pemetrexed-plus-platinum chemotherapy in both untreated and previously treated NSCLC patients with ALK-rearranged disease in advanced stage.24-25 Particularly, in previously-treated patients, PFS was significantly longer with crizotinib than with chemotherapy (median 10.9 months vs 7.0 months, respectively). 25
However, the responses observed in ALK-rearranged LUAD patients treated with crizotinib were limited to only 8-11 months, due to the rapid development of resistance mechanisms and to the relapse mainly observed at the level of central nervous system, due to the scarce capacity of crizotinib to bypass the blood-brain barrier.
In order to improve the inhibitory activity against rearranged protein and to treat brain metastases a second generation of more active ALK TKIs was developed.
Ceritinib
Ceritinib was evaluated in ALK-rearranged LUADs in comparison with standard chemotherapy: the ASCEND-04 phase III trial reported in untreated ALK-rearranged LUAD patients a significant improvement of median PFS in ceritinib-treated group compared to standard platinum-based chemotherapy (16.6 months vs 8.1 months, respectively). 26 The ASCEND-05 trial compared the efficacy and safety of ceritinib versus single-agent chemotherapy in patients with advanced ALK-rearranged NSCLC who had previously progressed following crizotinib and platinum-based doublet: ceritinib showed a significant improvement in median PFS compared with chemotherapy (5.4 months vs 1.6 months, respectively). 27 An indirect comparison of the clinical trials carried out using crizotinib or ceritinib as first-line treatment of ALK-rearranged LUAD patients showed a superiority of ceritinib in terms of PFS but not in terms of OS. 28
Brigatinib
Among patients with ALK-rearranged NSCLC who had not previously received any treatment with an ALK inhibitor, progression-free survival was significantly longer among patients treated with brigatinib than those treated with crizotinib. 28 Importantly, the rate of intracranial response was 78% and 29% in patients treated with brigatinib compared to those treated with crizotinib. 29 The results of this trial confirmed a highly significant effect of brigatinib in improving PFS in comparison with crizotinib (median PFS 24.0 vs 11.0 months, respectively). 30
Alectinib
The second generation ALK TKI alectinib was explored in comparison of other ALK inhibitors or of chemotherapy. The ALEX study randomized 303 ALK-rearranged NSCLC patients in first-line to receive either alectinib or crizotinib; alectinib administration resulted in a significantly longer PFS compared to crizotinib administration (25.7 months vs 11.1 months); furthermore, alectinib significantly decreased the events of central nervous system progression compared to crizotinib (12% vs 45%, respectively). 31 The evaluation of OS reported at one to five years for alectinib rates of 84%, 72%, 67%, 65% and 62.5%, compared for crizotinib of 82%, 65%, 57%, 51% and 45%, respectively; the data supported an OS benefit in alectinib arm. 32
The phase III ALUR study was a randomized trial of alectinib versus chemotherapy in advanced metastatic ALK-rearranged NSCLC patients previously treated with platinum-based chemotherapy and crizotinib; 72 patients were randomized to receive alectinib and 35 patients to receive chemotherapy. 31 Median PFS was 7.1 months with alectinib compared to 1.6 months with chemotherapy; in patients with measurable CNS (central nervous system) disease, the objective CNS response rate was 52% in the alectinib arm, compared to 0% in the chemotherapy arm. 33
The final results of the ALUR trial confirmed the superior PFS, ORR (overall response rate) and CNS ORR of alectinib versus chemotherapy; importantly, alectinib prolonged PFS versus chemotherapy in both patients with WT (wild-type) and TP53-mutant tumors: however, alectinib efficacy was considerably decreased in patients with TP53-mutated tumors. 34
Indirect treatment comparison of phase III trials involving either alectinib or brigatinib for front-line treatment of ALK-rearranged NSCLCs showed comparable OS between these two treatments. 35
ALK-rearranged LUAD patients treated with alectinib usually exhibit a characteristic tumor volume dynamic with initial response with tumor volume decrease, nadir and subsequent regrowth; the percent tumor volume changes, evaluated by serial computed tomography, induced by alectinib at nadir (-90.9%) were more pronounced in patients who were treated as first-line, compared to those treated in second-line. 36
Ensartinib
The eXalt3 phase II randomized clinical trial compared the response of 290 ALK-rearranged NSCLC patients to either ensartinib (another second-generation AKL-TKI) or crizotinib: patients treated with ensartinib displayed a significantly better PFS than those treated with crizotinib (25.8 months vs 12.7 months). The intracranial response rate was markedly higher in ensartinib than in crizotinib-treated patients (64% vs 21%, respectively). 37
Lorlatinib
Lorlatinib is a third-generation ALK TKI, highly potent, selective and brain-penetrant. Based on the results of a phase II study reporting an objective response rate of 47% in ALK-rearranged SCLC patients with advanced diseases with at least one exposure to ALK TKIs and with frequent CNS involvement, lorlatinib obtained accelerated FDA approval in the second and third-line setting. 38 The phase III randomized clinical trial CROWN compared in 296 SCLC patients with ALK rearrangement the therapeutic effect of lorlatinib with crizotinib: lorlatinib administration resulted in an improvement of PFS compared to crizotinib (not reached vs 9.3 months), in an increased ORR (76% vs 58%) and in an increased rate of intracranial objective responses (82% vs 23%). 39 An updated analysis of the results of the CROWN trial confirmed a durable benefit related to lorlatinib adminiastration: three-year PFS was 64% in the lorlatinib group and 19% in the crozitinib group; in patients without brain metastases, 1% of patients in the lorlatinib group and 23% in the crizotinib group had intracranial progression. 40
A phase II study carried out in ALK-rearranged NSCLC patients with specific CNS relapse showed a consistent therapeutic activity of lorlatinib, with 59% of intracranial objective responses and with a durable intracranial disease control. 41
A retrospective analysis in the CROWN study explored the potential clinical benefit of continuing lorlatinib administration beyond progressive disease (LBPD). 40 In both groups of patients A (those with previous crizotinib treatment) and B (those with at least one second-generation ALK TKI) the median overall survival was longer in patients who continued LBPD compared to patients who did not continue LBPD administration beyond progressive disease (LBPD). 42
Comparative analysis of the different ALK-TKIs
At time of writing, six different ALK TKIs have been approved for use as first-line treatment of advanced ALK-rearranged NSCLC (crizotinib, ceritinib, alectinib, brigatinib, ensartinib and lorlatinib); three of these TKIs, alectinib, brigatinib and lorlatinib are the most recommended. A comparative analysis of pharmacodynamic and clinical data supported a superiority of lorlatinib over the other ALK TKIs: (i) in vitro pharmacodynamic assays showed that lorlatinib is the most potent in inhibiting AKL-fusion proteins; (ii) pharmacokinetics studies showed that lorlatinib had the highest free drug fraction compared to other ALK TKIs (iii) lorlatinib achieved high cerebrospinal fluid concentration; (iv) the three-year PFS for lorlatinib was 63.5% compared to 46.4% and 43% for alcetinib and brigatinib, respectively; (v) for alectinib, a median PFS of 25.7 months was reached after a follow-up of 18.6 months, for brigatinib of 24.0 months after a median follow-up of 24.9 months and for lorlatinib not reached after a median follow-up of 36.7 months; (vi) lorlatinib significantly delayed intra-cranial time of progression compared to other ALK TKIs. 43
The analysis of the response related to the EML4-ALK variant type showed that in all clinical trials with alectinib, brigatinib and lorlatinib the PFS was shorter in patients with variant 3 EML4-ALK compared to those with variant 1 EML4-ALK; however, also concerning variant 3 EML4-ALK patients treated with lorlatinib displayed a longer PFS compared to those treated with alectinib and brigatinib (33.3 months vs 17.7 and 16.0 months, respectively).44-45
In line with the results of this comparative analysis, Ando and coworkers also, through a network meta-analysis of the most relevant phase III randomized controlled trials, reached the conclusion that lorlatinib was the most effective in prolonging PFS, followed by brigatinib, alectinib, ensartinib, ceritinib, crizotinib and chemotherapy. 46
Fourth-generation ALK-TKIs
New fourth-generation ALK-TKIs are under evaluation. Among these new inhibitors, the compound at the most advanced stage of development is AZP-3621 in phase III of evaluation in comparison with crizotinib in ALK-rearranged naïve NSCLC patients. Furthermore, the preclinical data showed a particularly promising pharmacodynamic profile of NVL-655, a new third generation ALK-TKI designed to remain active in patients who have developed resistance to current first-, second- and third generation ALK-TKIs and optimized for CNS penetrance. 47 TPX-0191 is a small macrocyclic compound designed to fit within the ATP binding pocket to inhibit ALK fusion proteins; its inhibitory activity against ALK resistance mutations is more pronounced than that of all approved ALK inhibitors. 48
Resistance to ALK-TKI
The benefit deriving from therapy with ALK-rearranged SCLCs with ALK-TKIs are strongly limited by the development of resistance mechanisms occurring after a variable period after starting the treatment. Resistance to ALK-TKIs can be distinguished into primary and secondary (acquired) resistance (Figure 2).
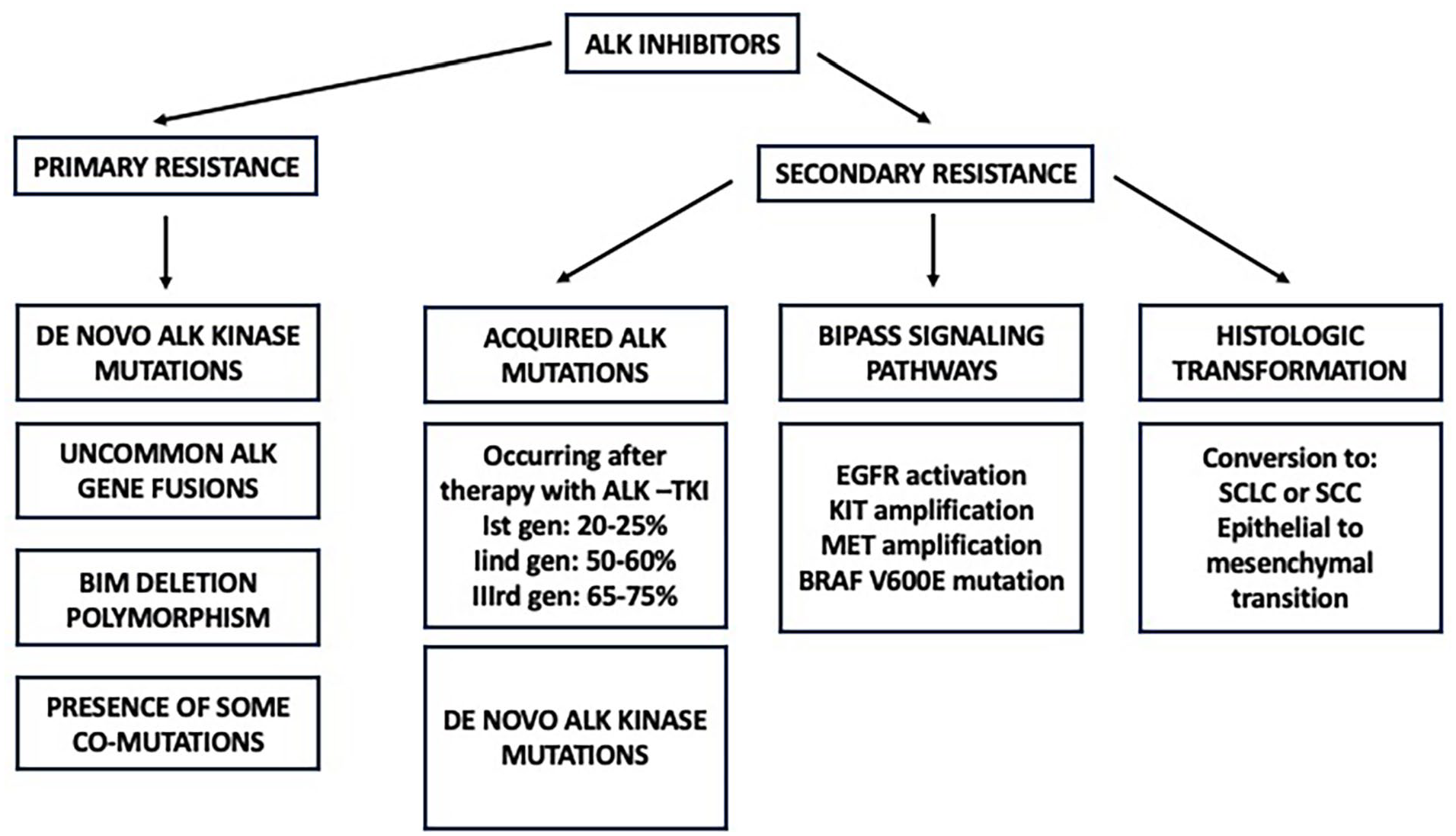
Mechanisms of resistance to ALK-TKIs, subdivided into primary and secondary mechanisms of resistance.
Primary resistance
Primary resistance can be defined as a resistance occurring from the beginning of the treatment with an ALK-TKI, with disease progression occurring within three months from the start of therapy.
Kang et al. in a group of 171 ALK-rearranged NSCLC patients reported the occurrence of primary resistance in 10.5% of these patients. 49 Among these resistant patients, 33% had uncommon ALK fusion partners, 22% had BIM deletion polymorphism, 11% PTEN/mTOR mutations and 5.5% a pre-existing ALK G3709A point mutation. 49 Wang et al. explored 92 ALK-positive NSCLC patients treated with crizotinib and showed that about 12% of these patients had primary resistance. Among these resistant patients, 18% had BIM deletion polymorphism, 9% PTEN or PIK3CA or CCND1 or SMARCA4 mutation, 9% CMTR1-ALK fusion and 9% a EML4-ALK fusion non A20. 50
Secondary (acquired) resistance
The mechanisms of acquired resistance to ALK-TKIs may be related to on-target and to off-target resistance mechanisms.
The main mechanism of on-target resistance is related to the generation of mutations that interfere with ALK inhibitors or with binding to kinase or of structural changes involving the kinase domain. ALK inhibitory point mutations are observed in about 30%, 50% and 70% of patients treated with first-, second- and third-generation ALK-TKIs.51-52 The most common ALK mutations observed in patients treated with ALK-TKIs are: L1196M, G1269A/S, C1156Y/T, G1202R, I1171T/N/S, S1206C/Y, E1210K, L1152P/R, F1174C/L/V, V1180L, I1151T and G1128A.51-52 One of these mutations, G1202R, is rarely observed after crizotinib treatment (2% of cases) but is frequently observed after treatment with second-generation ALK-TKIs (>20% of cases). 53 Lorlatinib is the only ALK-TKI active against G1202R mutation, as well as against frequent ALK point mutations such as L1196M and G1269A. 54
Off-target mechanisms are responsible of a significant part of resistance events occurring after ALK-TKI treatment, from 25% to 60% of cases. The main mechanisms of off-target resistance are related to activation of bypass signaling pathways, such as EGFR signaling, amplification of KIT or of MET and BRAF V600E mutation; morphological transformation of ALK-rearranged LUADs into small cell lung cancer or squamous cell carcinoma; epithelial-to-mesenchymal transition. Some recent review papers have analyzed in detail the impact of these off-target resistance mechanisms in the clinical resistance to ALK-TKIs.55-56
Conclusion
The studies carried out in the last two decades have shown that the introduction of target therapy consistently improved the survival of ALK-rearranged LUAD patients. The development of three generations of ALK inhibitors has shown that more potent and specific is the ALK-TKI, better clinical benefit can be obtained. The data until now observed for lorlatinib have shown after a follow-up of three years a PFS not yet reached. The updated long-term data support the superiority of lorlatinib over other ALK-TKIs and suggest the lorlatinib could represent the gold standard for first-line ALK-positive NSCLC patients. Fourth generation ALK-TKIs are expected to still improve this therapeutic scenario.
In spite these significant improvements, many challenges remain unresolved in the treatment of ALK-rearranged NSCLC, such as the existence of primary resistance in a minority of patients or the development of secondary resistance due to in-target and off-target mechanisms and future studies will attempt to define strategies to bypass these events.
These findings strongly support the need for a full molecular characterization of these tumors in addition to the identification of ALK rearrangement for an optimal prognostic evaluation and for definition of an optimal individual therapeutic strategy.
Footnotes
Author Contributions
Conceptualization and literature research UT, GC and EP; writing-original draft preparation, UT, GC and UT; manuscript editing, UT.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.