
Abstract
Introduction:
The H3K27M-mutant diffuse midline glioma (DMG) was first included in the World Health Organization (WHO) Classification of central nervous system (CNS) tumors in 2016, and confirmed in its fifth edition. The biological behavior and dismal prognosis of this tumor resemble diffuse intrinsic pontine gliomas (DIPG). Homogeneously-treated series are rarely reported.
Methods:
From 2016 onwards, we treated patients with DMG with radiotherapy and concomitant/adjuvant nimotuzumab/vinorelbine, plus re-irradiation at relapse, as already done for DIPG.
Results:
We treated nine patients, seven females, with a median age at diagnosis of 13 years. Tumor sites were: thalamic in five cases, pontocerebellar in two, pineal in one, and paratrigonal with nodular/leptomeningeal dissemination in one. Three patients were biopsied, and six had partial tumor resections. Central pathological review was always performed. The median time to local progression was 12.7 months, and the median overall survival was 17.8 months. Six patients died of tumor progression, one of cerebral bleeding at progression. Two were alive, one in continuous remission, the other after relapsing, at 38.6 and 46.3 months after diagnosis. Progression-free survival was 33.3% at one year. Overall survival was 88.9%, 33.3% and 22.2% at 1, 2 and 3 years, respectively.
Conclusions:
This is a small series of homogeneously-treated DMG patients. The results obtained are comparable with those of DIPG patients. Given the phenotypically- and molecularly-defined setting of DMG and severe outcome in this orphan population, they should be treated and included in registries and protocols of DIPG.
Introduction
Pediatric high grade glioma (HGG; WHO grade III-IV), and diffuse intrinsic pontine glioma (DIPG) in particular, pose one of the great challenges in pediatric oncology, carrying the highest mortality in this patient population. 1 The H3K27M-mutant diffuse midline glioma was first included in the 2016 WHO Classification of CNS tumors as a distinct grade IV glioma 2 and it was confirmed in the latest 2021 edition. 3 Recent studies4-6 found that, regardless of its morphological features, this tumor subtype has an aggressive clinical behavior and a dismal prognosis, sharing the same outcomes as DIPG. The thalamus, pineal region, brainstem and spinal cord are the typical sites of origin of DMG, but the presence of the H3K27M mutation seems to be a negative prognostic indicator in pediatric HGG, whatever the site involved. 5
The tumor cells contain a missense mutation caused by a lysine-to-methionine substitution at amino acid position 27, in either the HIST1H3B (H3.1) or the H3F3A (H3.3) histone variant. The mutation results in significant epigenetic reprogramming, which contributes to tumorigenesis and gives rise to a tumor phenotype with genetic, epigenetic and clinical features that differ from its wild-type counterpart and from other non-midline HGG. Both histone variants (H3.1 and H3.3) can be tested with CLIA–certified DNA sequencing or immunohistochemical staining. 6
Genome analysis of K27M-mutant DMG has revealed a number of cooperating genetic alterations including: TP53 overexpression, ATRX loss, and none of the genetic alterations generally associated with a more favorable prognosis, such as IDH1/2 mutations or 1p/19q co-deletion.7,8 DMGs have rarely been found to harbor both the histone H3-K27M and the BRAF-V600E mutation.6,9 DMGs account for 10-20% of all pediatric brain tumors, with a median age at presentation ranging between six and nine years.
Due to the rarity of reports on their ad-hoc treatment to date, we describe our homogeneously-treated series of nine patients.
Materials and methods
All patients were referred at our Institution (Fondazione IRCCS IStituto Nazionale dei Tumori, Milan) for adjuvant treatment after surgery performed elsewhere. Resections were classified as complete, subtotal (removal of >90% of the tumor) or partial (removal of >50% but <90%), while resections of <50% of the tumor were classified as non-debulking, including biopsies. 5 Central neuropathological review was performed according to standard Italian procedure, which is referring cases to the Neuropathology Unit at La Sapienza University in Rome. A central histopathological diagnosis of DMG according to the WHO 2016 Classification was necessary to include the patients.
All patients with DMG diagnosed between April 2016 and November 2018 were staged and treated in the same way. Tests performed within 14 days prior to any therapy included: cranial and spinal magnetic resonance imaging (MRI) with gadolinium (to identify the index lesion); full clinical and neurological examination, recording body weight and height, Karnofsky score or Lansky performance status; full blood counts, renal, kidney and coagulation parameters. Response to treatment was assessed using RECIST criteria, 10 considering the diameters measured on T2-weighted images.
Systemic treatment consisted of vinorelbine 20 mg/m2 with nimotuzumab 150 mg/m2 administered weekly during the first 12 weeks of treatment. Focal radiotherapy was generally delivered from weeks 3 to 9, for a total dose of 54 Gy (induction phase). Vinorelbine 25 mg/m2 and nimotuzumab (same dosage) were given every other week thereafter (maintenance phase) until the tumor progressed, or for up to two years. In cases of local progression, re-irradiation consisted of 19.8 Gy, fractionated over 11 days (Figure 1). In cases of progression with tumor dissemination, craniospinal irradiation (CSI) doses and schedules varied, depending on the sites of disease already irradiated, but the total dose was generally 36 Gy, delivered with daily fractions of 1.8 Gy.
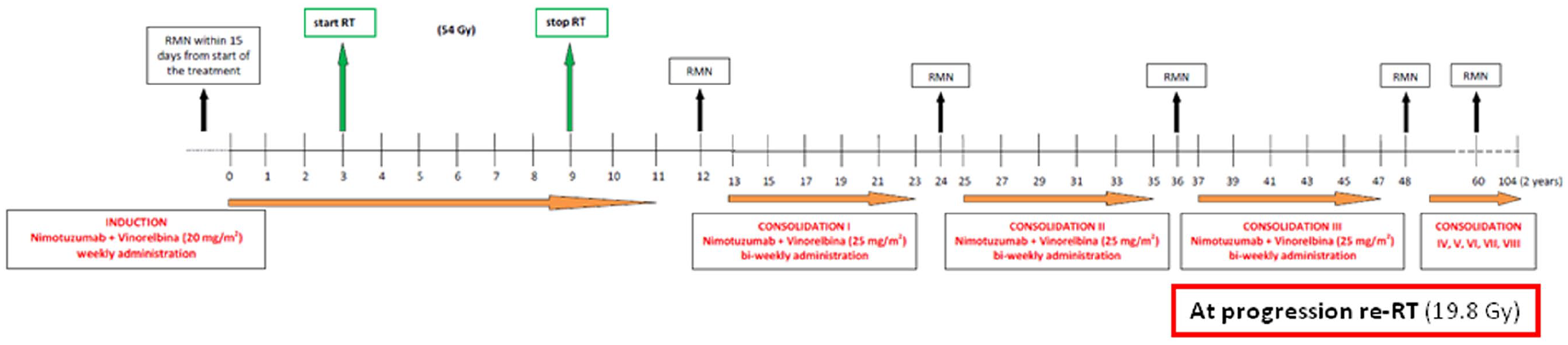
Treatment flow-chart for patients with DMG.
MRI was repeated every 12 weeks throughout the treatment, or sooner if symptoms worsened or pseudoprogression was suspected.
Immunohistochemistry for detecting the H3.3 mutation
Formalin-fixed and paraffin-embedded material was available for all cases. Immunohistochemical studies were performed using a Leica BondMax immunostainer with the following antibodies: rabbit polyclonal anti-histone-H3K27M antibody (#ABE419 Millipore, 0.5 ug/ml, 1:1000); and rabbit monoclonal anti-trimethyl-histone-H3 (Lys27) antibody (C36B11, Cell Signaling Tech, 1:200). Slides were independently scored for H3K27M and H3K27me3 staining by two neuropathologists. Samples were considered positive when tumor cells showed strong nuclear staining for H3K27M, and non-neoplastic elements (such as endothelial cells or lymphocytes) were negative. H3K27me3 immunoreactivity and H3K27M positivity were mutually exclusive. Positive K27M samples did not stain for H3K27me3.
Statistical analyses
The main endpoints of the study were overall survival (OS) and progression-free survival (PFS). Secondary endpoints were post-relapse survival (PRS) and local relapse-free survival (LRFS). Survival was calculated from the date of the first surgical procedure leading to the diagnosis for OS, PFS and LRFS, and from the date of first recurrence for PRS. OS and PRS were defined as time to death due to any cause, and censored at the latest follow-up for patients still alive. PFS was defined as the time to any radiological or clinical progression or death due to any cause (whichever came first), and censored at the latest follow-up for patients still alive and progression-free. LRFS was defined as the time to the first local recurrence, and censored at the time of dissemination or death due to any cause (whichever came first). All survival probabilities were estimated with the Kaplan-Meier method. The median follow-up was estimated on OS data with the reverse Kaplan-Meier method.
The protocol adopted was approved by the Institutional Review Board (IRB) as 149/18 and patients’ parents, or patients over 18 years old signed a consent form before receiving treatment.
Results
Neuropathological assessment
The first pathological diagnoses (performed elsewhere, before referral for adjuvant treatment) were: glioblastoma in four cases, anaplastic astrocytoma, diffuse astrocytoma and glial tumor with oligodendroglial features in one case each and DMG (H3K27M mutant) in two cases. Central pathology review revealed H3.3 K27M mutation on immunohistochemical staining in all nine cases. The Ki67 value varied, ranging between 2% and 70%. No ATRX loss was seen in five cases tested. Four samples were found positive for the Tp53 mutation, while five tested negative. The BRAFV600E mutation was identified in one of seven patients tested with PCR sequencing. FISH for TRK1/3 fusions was negative in four patients tested. IDH1-2 mutations were negative in three patients tested. An EGFR mutation was found in the patient with bithalamic tumor. 11
Patients’ characteristics
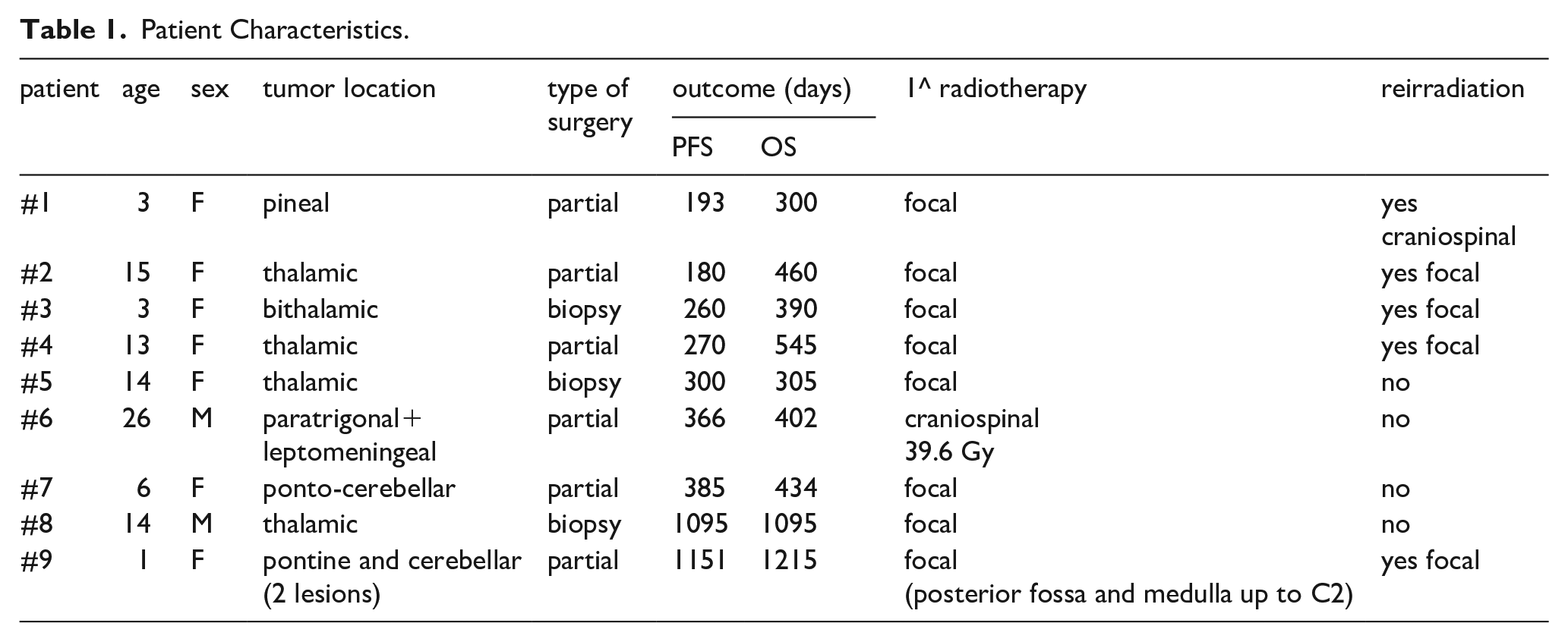
Nine consecutive patients with DMG were diagnosed and treated from May 2016 to December 2018. Seven were females. The median age at diagnosis was 13 years (range 1-26). The median duration of symptoms at the time of diagnosis was 40 days (range 10 days to 4 months). Eight patients had localized disease, while one had nodular and leptomeningeal metastases at diagnosis. Five patients with localized disease had thalamic tumors (bithalamic in one), two had tumors in the pontocerebellar structures (one with two distinct lesions), one had tumor in the pineal gland, and the paratrigonal area was involved in one (with nodular and leptomeningeal metastases).
Six patients had their tumors partially resected, while three only had biopsies (Table 1). The eight patients with localized tumor all completed the prescribed radiotherapy (total dose 54 Gy). The patient with disseminated disease received 39.6 Gy of CSI fractioned over 22 days. Response after the induction phase was stable disease or a minor volume reduction in five patients and possible pseudoprogression in the other four, who were reassessed with MRI after 45 days, with tumor dissemination found in one case and stable disease in three. One of the patients with pseudoprogression (and disseminated tumor at diagnosis) concomitantly developed hydrocephalus and needed a ventriculo-peritoneal shunt.
Patient Characteristics.
With a median observation period of 42.5 months (1st and 3rd quartiles: 38.6-46.3 months), the PFS and OS (95%CI) at 1 year were 33.3% (13.2-84.0%) and 88.9% (70.6-100), respectively. At 2 and 3 years, the OS was 33.3% (13.2-84.0%) and 22.2% (6.6-75.4%), respectively (Figure 1). No patients died of toxicity (Figure 2).
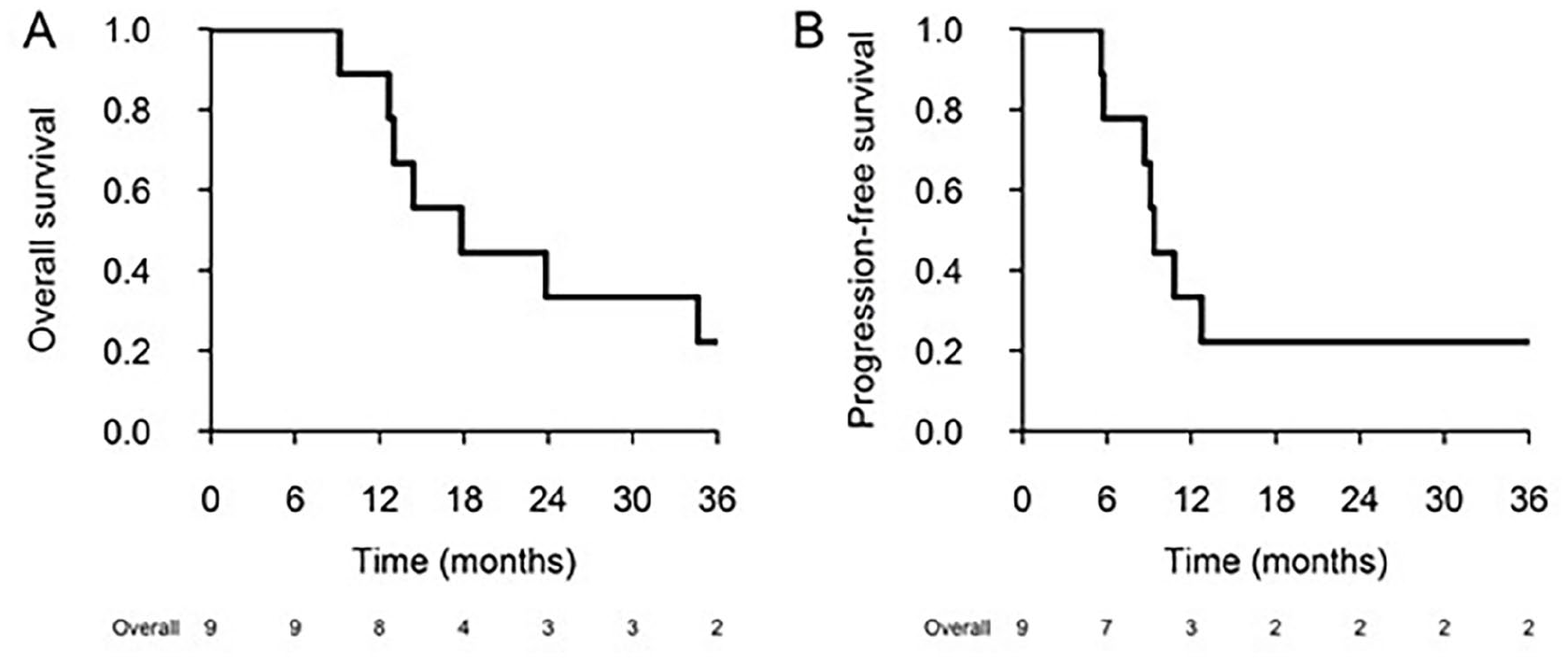
Overall survival (OS) and Progression free survival (PFS) across the patient cohort. In the x-axis the detailed number of alive patients.
Six patients had local progressions after a median of 12.7 months (range 5.6-38.6 months) from diagnosis. Three were re-irradiated with a total dose of 19.8 Gy; one relapsing with disseminated tumor had CSI, receiving a total dose of 32.4 Gy delivered in 18 days. Among the other patients with progressive disease, two underwent re-surgery at other centers, and were then treated with second-line chemotherapy, one developed a cerebral hemorrhage after radiological evidence of progressive disease, and one (treated with CSI at diagnosis) died during second-line chemotherapy two months after disease progression. Hydrocephalus requiring a shunt was present at diagnosis in five patients, while it developed during treatment in one, and after tumor progression in one. The patient with BRAF-V600E mutation progressed six months after starting the treatment and further progressed after re-irradiation, she was then hospitalized due to deteriorating neurological conditions. She showed a rapid clinical and radiological response to dabrafenib and trametinib association, administered for compassionate purposes, which persisted for four months. One patient refused further treatment after six months, when her tumor was stable. Median survival after relapse was 6.4 months (range 1.7-25.6 months).
Two patients are long-term survivors. One was diagnosed at 20 months of age with two tumor localizations, one bulbo-pontine and the other (which was completely removed) cerebellar hemispheric. After radiotherapy, a reduction in the residual tumor persisted all along the two years of treatment, then a local relapse was observed after 38.6 months. Tumor control was achieved again with partial surgery, re-irradiation and the same systemic treatment. The other patient, with hereditary neurofibromatosis type 1, was diagnosed with thalamic disease at 14 years old. After completing the two years of treatment with stable disease, he is in good clinical conditions 38.6 months after primary surgery.
Toxicity
Hematological profile of safety of this regimen has been already reported elsewhere in DIPG protocol. 12 However no patient delayed or modified treatment because of hematological or other toxicities. The cases of pseudoprogression, hydrocephalus and cerebral hemorrhage have already been described above; all these events were surely not drug-correlated.
Discussion
H3K27M-mutant DMG carries a very grim prognosis. Overall survival is typically 7-11 months in children, and 8-19 months in adults. 6 Maximal safe resection is recommended, if feasible, but the role of surgery is often only diagnostic, radical removal being impossible due to the tumor’s critical location. No standard therapy has been clearly identified as yet, but patients are generally given the same focal radiotherapy as for all aggressive gliomas.13,14 While the effects of radiotherapy seem to be very short-lived, the role of chemotherapy still appears to be marginal. Temozolomide is indicated for adult glioblastoma, and still used as standard in pediatric patients as well. It seems to have no effect, however, partly because H3K27M-mutant gliomas are all MGMT(O 6 -methylguanine-DNA methyltransferase)-unmethylated, and therefore probably all resistant. 15 Over an accrual period of two years, we adopted the same strategy as for DIPG 12 for the few DMG patients referred to us. Having obtained results that compare favorably with those reported in previous trials5,16 we have now included non-DIPG DMG patients in our current ongoing protocol as well (EudraCT Number: 2015-002185-23). One of these was a multicentric randomized trial16,17 designed for newly diagnosed pediatric high grade glioma that demonstrated a clear negative impact on survival for subgroup of 24 patients with midline K27M tumors [median event free survival (EFS)/OS: 7.9 and 14.2 months respectively]. The other was a large retrospective series 5 on diffuse midline glioma K27M-mutant (including DIPG) taken from German HIT-HGG registry that extrapolated data on the thalamic DMG cohort (24 patients) showing one year EFS and OS of 21 and 63% respectively. While there were no toxic deaths in our series, the treatment was associated with pseudoprogression in nearly half the patients after the first course of radiotherapy. When using target therapies like the anti-EGFR nimotuzumab, response to radiation may be associated with tumor cell death, immune activation, and edema, leading to pseudoprogression. 18
In line with some previous reports,19,20 more than 40% of our patients (four of our nine patients) expressed Tp53-mutant tumors. This would seem to suggest a link with the severe prognosis for cases of DMG, but this remains to be seen in larger studies.
Some papers describe a favorable prognostic effect of pseudoprogression that we were unable to examine in our small series. 21 We also noted a relatively high ventricular shunting rate in our series, due to the large, deep-seated lesions involved, and the frequent tumor dissemination at both diagnosis and relapse, making our patients particularly fragile both at diagnosis and during the course of their disease. Re-irradiation nonetheless proved just as feasible in this subgroup of DMG patients as in cases of DIPG, 12 and is a second-line therapy always worth considering.
The identification of the H3K27M mutation in DMG and DIPG has prompted the testing of new potential target drugs in ongoing clinical trials. The hypomethylation associated with the H3K27M mutation suggests a correlation between chromatin dysregulation and malignancy in pediatric HGG. Novel agents that target the processes involved in epigenetic regulation are now under investigation, including histone deacetylase (HDAC) inhibitors like panobinostat and BET brododomain inhibitors.22,23,24 A phase 1 trial is currently examining the safety of administering panobinostat in children and young adults with DIPG, HGG and medulloblastoma (PBTC-047). Another ongoing phase 1 trial focuses on combinations with vorinostat and temsirolimus in children and young adults with DIPG (Eudract NCT02420613). ONC201 is a small molecule selective antagonist of dopamine receptor D2/3 (DRD2/3) that has shown anticancer activity in preclinical models of K27 mutant HGG. 25 DRD2 expression is highest in midline structures of the brain. Few reports26,27 have described ONC201 as a potential target for DMG in recurrent adult and pediatric setting (including DIPG). A phase I study of oral ONC201 in pediatric patients with newly diagnosed DIPG and recurrent/refractory H3K27M gliomas is actually ongoing (Eudract NCT03416530).
Convection-enhanced delivery (CED) is also being studied as an attractive means of delivering therapeutics directly to DMG, as it bypasses the blood-brain barrier and enables the treatment of a relatively large volume of tissue with small amounts of infusate. 28 A recent study found that cell cultures from patients’ H3K27M-mutant gliomas strongly expressed disialoganglioside (GD2) and, in cultured cells, anti-GD2 CAR T cells revealed a significant antigen-dependent cytokine production and killing of H3K27M-mutant DIPG cells that was not seen in a H3K27M-wild type cell culture. Anti-GD2 CAR T cells proved effective in orthotopically engrafted H3K27M tumors, although a significant toxicity was observed in treated mice. 29 Whether this strategy is applicable to patients and sustainable remains to be seen. The co-occurrence of histone H3K27M and BRAF V600E mutations had already been described, 9 but is extremely rare in DMG. BRAF V600E mutation inhibitors should nonetheless be used when indicated. While the prospects of new findings emerging and being rapidly applied in clinical practice is exciting, we feel that our strategy – which has led to two of our nine patients achieving a remarkably long survival - remains an acceptable option until such time as the results of other trials become available.
Table of abbreviations
CART cells chimeric antigen receptor T cells
CED convection-enhanced delivery
CNS central nervous system
CSI craniospinal irradiation
DIPG diffuse intrinsic pontine gliomas
DMG H3K27M-mutant diffuse midline glioma
EFS event free survival
HGG high grade glioma
IRB institutional review board
LRFS local relapse-free survival
MGMT O 6 -methylguanine-DNA methyltransferase
MRI magnetic resonance imaging
OS overall survival
PFS progression-free survival
PRS post-relapse survival
Footnotes
Acknowledgements
We thank Lega Italiana per la Lotta contro i Tumori (LILT Milano Monza Brianza) for continuous assistance of patients and family during treatment.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partially supported by the Italian Ministry of Health and by Regione Lombardia (project ID NET-2019-12371188 to Maura Massimino).