
Abstract
Ayme-Gripp syndrome is a rare genetic disorder caused by mutations in the musculoaponeurotic fibrosarcoma gene. This report presents a detailed case of a 12-year-old boy with Ayme-Gripp syndrome who was found to carry a pathogenic c.176C>A (p.Pro59His) variant in the musculoaponeurotic fibrosarcoma gene. His clinical phenotype included the most severe short stature documented to date as well as previously underreported features such as epileptic seizures, in addition to the classic findings of hearing loss, intellectual disability, and craniofacial/skeletal malformations. The patient’s epilepsy was effectively managed with sodium valproate, alongside comprehensive supportive care. This case highlights the broad phenotypic variability associated with musculoaponeurotic fibrosarcoma gene mutations and underscores the importance of deep phenotyping in expanding our understanding of ultra-rare disorders such as Ayme-Gripp syndrome.
Keywords
Introduction
Ayme-Gripp syndrome (AYGRPS, OMIM 601088) is a congenital disorder caused by pathogenic variants in the musculoaponeurotic fibrosarcoma (MAF, OMIM 177075, NM_005360.5) gene, which is located at chromosome 16q23.2. With fewer than 35 cases reported worldwide, the syndrome exhibits considerable mutational heterogeneity. It is characterized by sensorineural hearing loss, neurological abnormalities (e.g. intellectual disability, epileptic seizures, and global developmental delay), ocular manifestations (e.g. congenital cataracts), and craniofacial and skeletal malformations. Owing to its multisystem involvement, abnormalities in the cardiovascular, renal, and endocrine systems may also occur, presenting as pericarditis, glomerulonephritis, and hypothyroidism. These features may overlap with the phenotypes of Down syndrome. 1 The rarity of AYGRPS, combined with its diverse and complex manifestations, poses challenges for diagnosis and treatment.
Despite previous characterizations, the full clinical scope of AYGRPS remains incompletely defined, particularly regarding the extent of phenotypic variability and genotype–phenotype correlations. Herein, we report a comprehensive clinical profile of a 12-year-old boy diagnosed with AYGRPS caused by a missense variant (c.176C>A, p.Pro59His) in the MAF gene, who presented with the most severe short stature reported to date in AYGRPS and exhibited previously underrecognized features, including epicanthal folds and pectus excavatum. Through detailed clinical profiling and a comprehensive review of the literature, we aim to refine the phenotypic spectrum of AYGRPS, elucidate genotype–phenotype associations, and contribute to the growing case database, with implications for improved diagnostic accuracy and future research directions.
Case presentation
A 12-year-old boy was recruited from the Children’s Welfare Center. The birth history and parental anthropometrics were unavailable for review. At the age of 2 years, he was sent to a local social welfare center. Admission examination showed a weight of 12 kg, a height of 76 cm (−3.2 standard deviation (SD)), a head circumference of 43 cm (−2.8 SD), and a chest circumference of 45 cm. Other physical examination findings included pectus excavatum, malformed ears, typical dysmorphic facial features, and bilateral testicular swelling. The anterior fontanelle had already closed. However, he was unable to speak, remained in a passive posture, and lacked the ability to sit, stand, or walk.
Although detailed sequential medical records were unavailable, two isolated growth measurements indicated his profound growth failure: his height was 83 cm at the age of 3 years and 92 cm at the age of 5 years, both far below the 3rd percentile for his age and consistent with the severe postnatal growth retardation characteristic of AYGRPS. During this period, he received basic nursing and nutritional support but had no access to formal physical, occupational, or speech therapy. Caregiver reports and sparse institutional notes consistently indicated a static and severe global developmental delay, with no acquisition of speech, independent sitting, standing, or walking.
At 5 years and 11 months of age, he was referred to the Children’s Hospital of Zhejiang University School of Medicine due to recurrent cyanosis over a 4-month period. Clinical manifestations included sudden-onset cyanosis of the lips without obvious triggers, accompanied by frothing at the mouth, limb weakness, and loss of consciousness. Approximately six episodes occurred during this period. Based on these characteristics and examination results, epilepsy was considered, and topiramate was administered orally twice daily. At the age of 11 years, to improve symptom control, the medication was adjusted to sodium valproate (0.1 g twice daily), resulting in sustained control with no recurrence to date. Despite severe intellectual disability and complete dependence for daily living, he had a gentle temperament and received in-home special education services.
Current physical examination showed a height of 121 cm (−3.52 SD) and a weight of 15.5 kg (−4.56 SD). Distinctive facial features included prominent frontal bones, high forehead, midface hypoplasia, low-set ears, hypertelorism with epicanthal folds, a flattened nasal bridge, long philtrum, and a small mouth with thin upper lips (Figure 1). The patient also presented with finger deformities, and the previously noted pectus excavatum persisted. There were no positive findings of a single simian crease, hepatosplenomegaly, lymphadenopathy, or history of recurrent infections. Muscle tone was normal. On gross clinical examination, he demonstrated markedly diminished responses to visual stimuli and sound. However, formal ophthalmologic evaluation (including slit-lamp examination) and objective audiometric testing (such as auditory brainstem response) were not performed due to severe disability and institutional care constraints. Therefore, the precise nature of the sensory impairments (e.g. cataracts and sensorineural hearing loss) could not be definitively established. He was unable to communicate or follow commands and had significantly reduced limb muscle strength with urinary and fecal incontinence. Chewing function was impaired, requiring a semi-liquid diet, and a corrective chair was used for daily support.
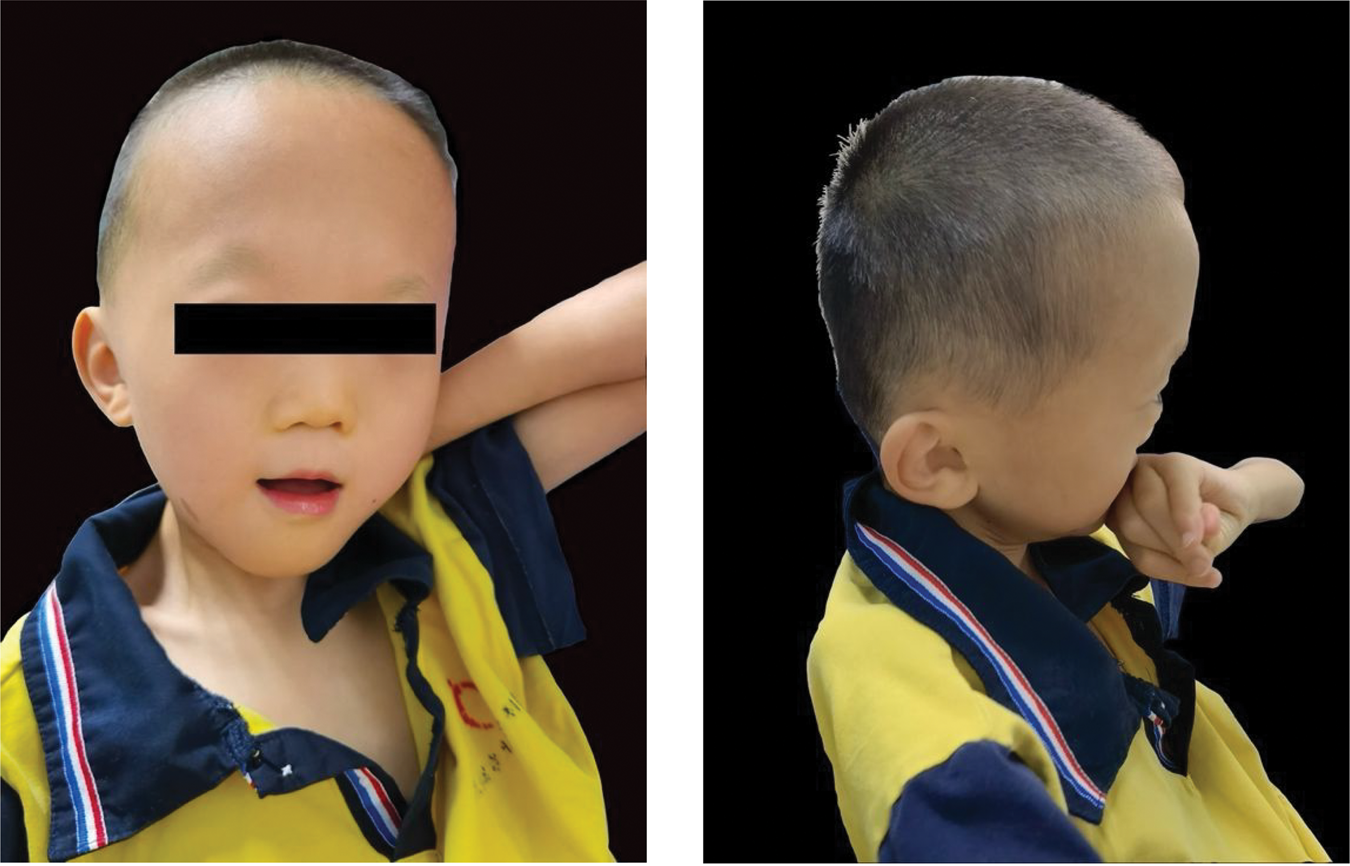
Facial characteristics of a pediatric patient with AYGRPS caused by a heterozygous MAF variant. Front view demonstrating brachycephaly, high forehead, flattened nasal bridge, long philtrum, small mouth, and thin upper lip. Lateral profile highlighting a prominent frontal bone, midface hypoplasia, and low-set ears.
Cranial magnetic resonance imaging showed flow-void vascular shadows in the left basal ganglia region, suggestive of a developmental venous malformation. No significant abnormalities were found on video electroencephalography, cardiac ultrasound, or abdominal ultrasound.
Genetic analysis
Genetic identification and analysis
Genomic DNA was extracted from peripheral blood at the Children’s Hospital of Zhejiang University School of Medicine in Hangzhou in August 2023 and was subjected to whole exome sequencing, which identified a heterozygous missense variant in the MAF gene. Sanger sequencing provided direct validation of the variant, as shown in Figure 2. At the genomic level, the mutation is designated as chr16:g.79633624C>A; it corresponds to NM_005360.5:c.176C>A at the complementary DNA level and results in the amino acid substitution p.(Pro59His).

Sanger sequencing. The c.176C>A mutation in the MAF gene validated by Sanger sequencing. The upper panel shows forward sequencing, and the lower panel shows reverse sequencing. MAF: musculoaponeurotic fibrosarcoma.
Variant interpretation and pathogenicity assessment
Based on the results of prediction tools such as SIFT, PolyPhen-2, and FATHMM v2.3, the pathogenicity of this missense variant was predicted using the REVEL algorithm. According to the American College of Medical Genetics and Genomics (ACMG) guidelines, this variant was classified as likely pathogenic, confirming the diagnosis of AYGRPS.
Structural modeling
Structural analysis using PyMOL revealed that a rigid proline was substituted by a polar histidine at position 59 within the N-terminal transactivation domain of MAF (UniProt: O75444), as illustrated in Figure 3.
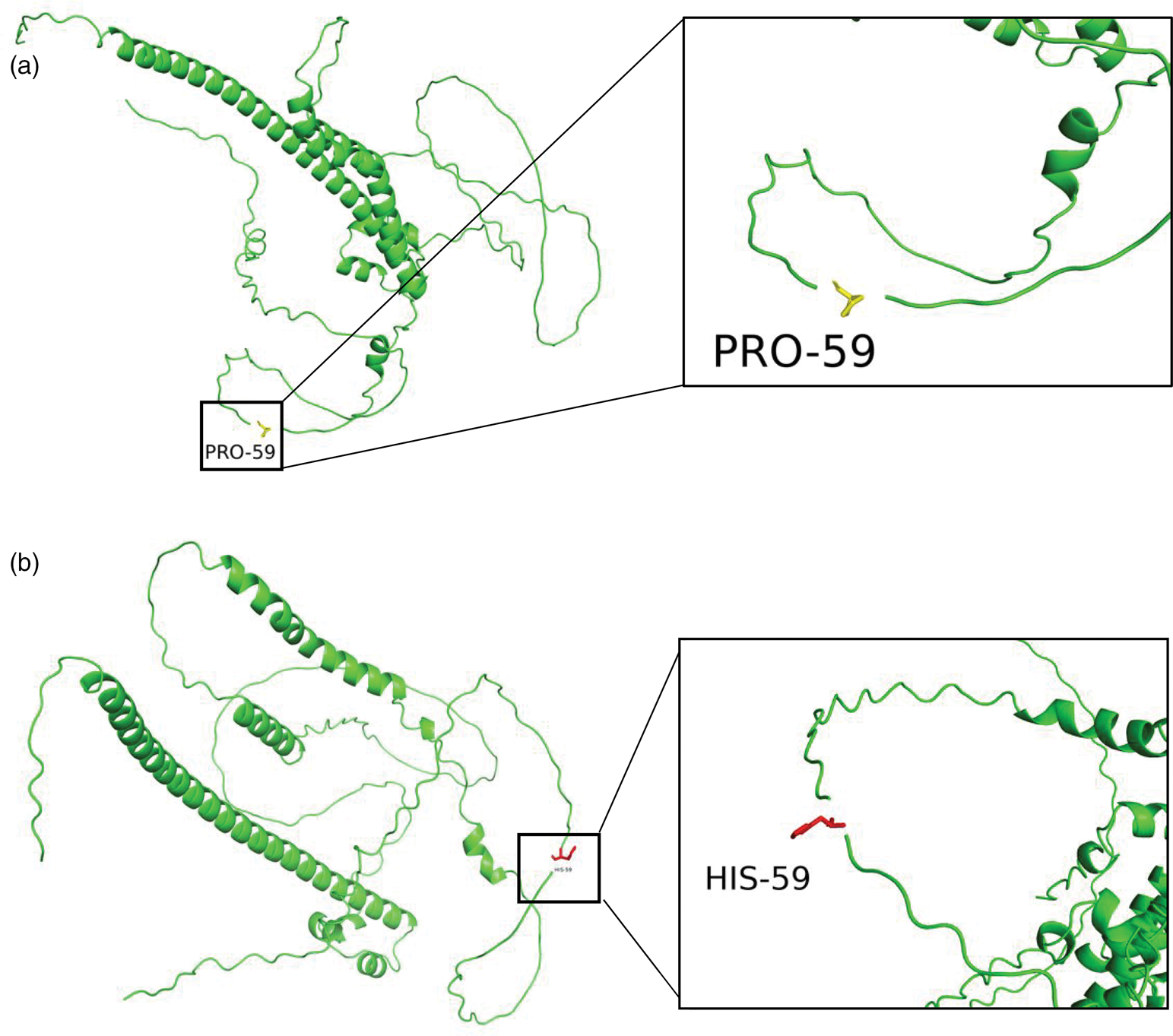
Wild-type (a) and mutant (b) MAF protein structures. The mutated residue (Pro59→His59) is shown in yellow (wild-type) and magenta (mutant type) sticks. MAF: musculoaponeurotic fibrosarcoma.
Currently, the child continues to require multidisciplinary management, with a focus on monitoring cardiac function and neurodevelopmental progress.
The reporting of this study conforms to Case Report (CARE) guidelines, and all patient details have been de-identified to protect anonymity. 2
Discussion
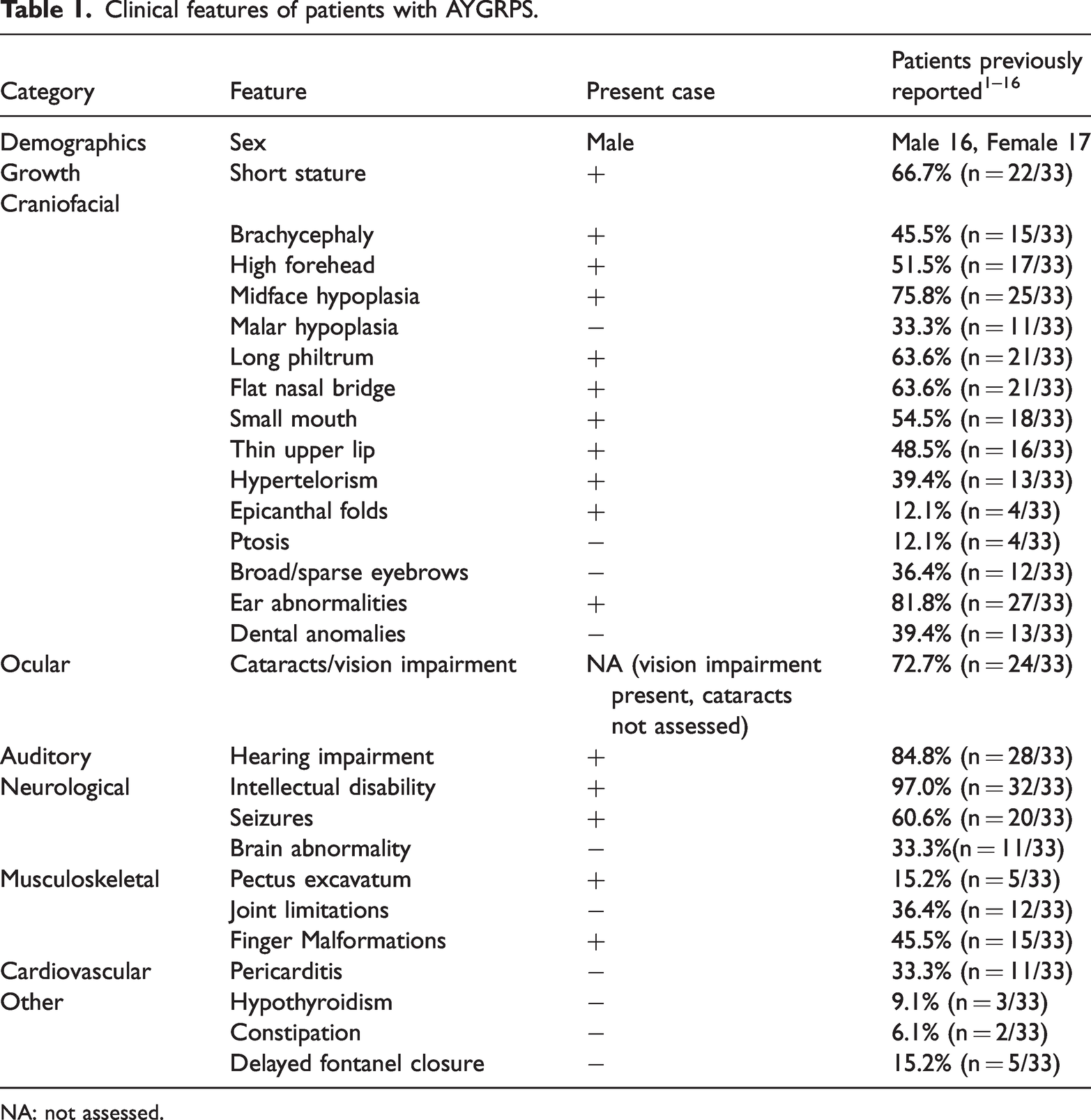
AYGRPS is an exceedingly rare genetic disorder, with fewer than 35 cases reported worldwide to our knowledge. Our pediatric case exhibited characteristic features, including reduced height (−3.52 SD), brachycephaly, prominent frontal bone, high forehead, midface hypoplasia, low-set ears, hypertelorism, epicanthal folds, flat nasal bridge, long philtrum, small mouth, thin upper lip, intellectual disability, finger malformations, and epileptic seizures, along with impaired vision and hearing (Figure 1). To contextualize our patient’s phenotype, we systematically reviewed the literature and compiled data from 33 previously reported genetically confirmed cases of AYGRPS. A comparison between this literature cohort and our present case is summarized in Table 1.1,3–17 Among the reported clinical features, the most common include intellectual disability, seizures, reduced height, typical facial features (midface hypoplasia, long philtrum, ear abnormalities, and flat nasal bridge), vision impairment, and hearing loss, all of which were present in our case. Due to practical constraints related to institutional care and the patient’s severe disability, comprehensive ophthalmologic and audiologic evaluations could not be performed. Therefore, although the observed poor visual and auditory responses were clinically compatible with the sensorineural deficits frequently reported in AYGRPS (most commonly cataracts and sensorineural hearing loss), these specific diagnoses remained unconfirmed in our patient. This case highlights the challenges of fully characterizing the phenotype in similar social welfare settings. Notably, epicanthal folds and pectus excavatum observed in our patient were relatively uncommon manifestations. It is important to emphasize that although this case shares the feature of short stature with many reported cases, it represents the most severe short stature documented in AYGRPS to date (−3.52 SD), as the previously most affected case had an SD of −3.48. 10 Although AYGRPS shares macro-features such as intellectual disability, short stature, and distinctive facial features with Down syndrome, key distinguishing features in our case include the absence of simian creases, protruding tongue, and atrioventricular septal defects (common in Down syndrome), together with the presence of a high forehead, severe midface hypoplasia, and a long philtrum (more typical of AYGRPS). Furthermore, sensorineural hearing loss in AYGRPS contrasts with the predominantly conductive hearing loss observed in young individuals with Down syndrome. 18 Therefore, in children presenting with developmental delay and dysmorphic facial features, AYGRPS should be considered in the differential diagnosis alongside Down syndrome, and genetic testing is recommended to establish a definitive diagnosis.
Clinical features of patients with AYGRPS.
NA: not assessed.
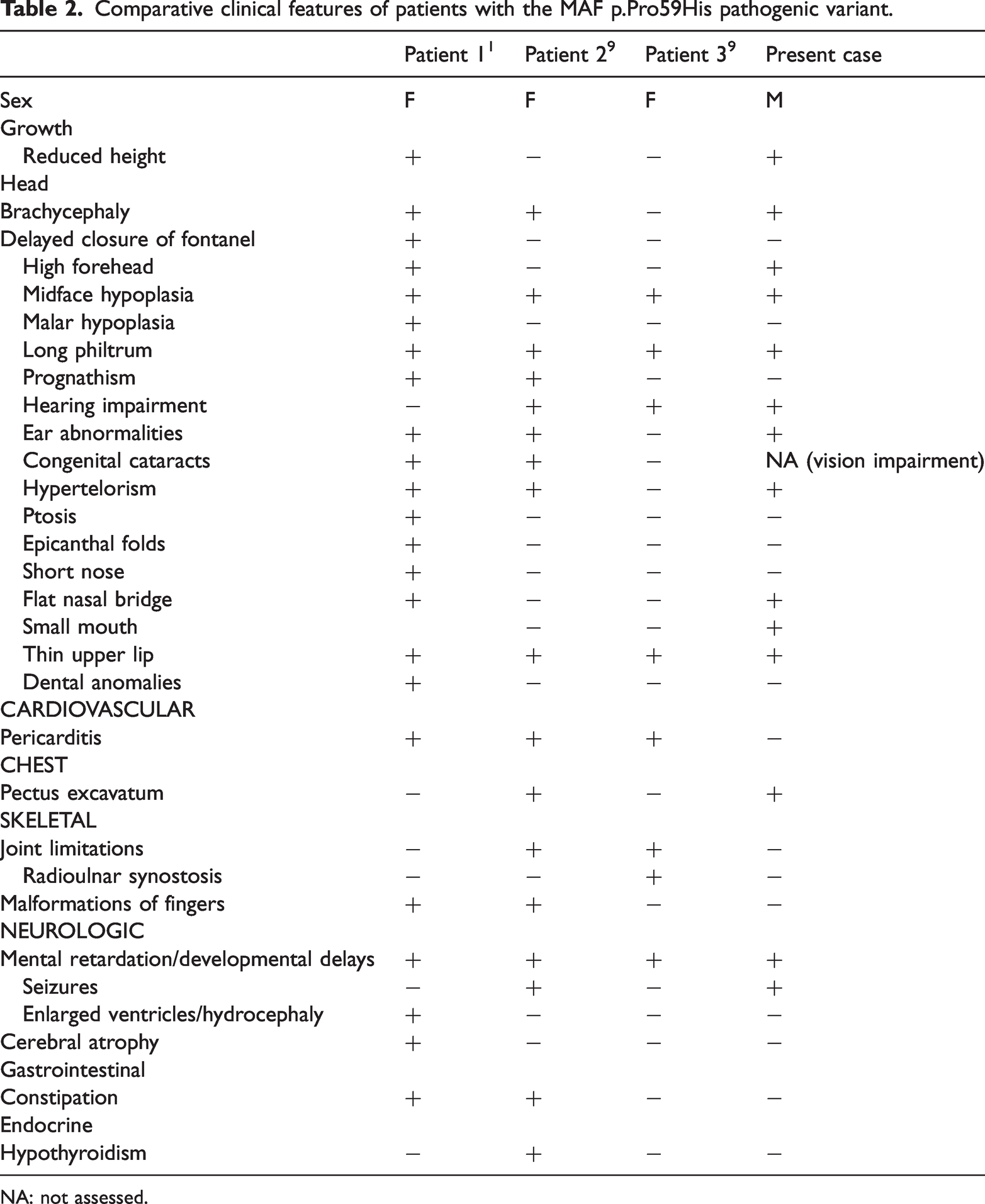
Genetic testing identified a c.176C>A (p.Pro59His) pathogenic missense variant in the MAF gene (Figure 2), which was classified as likely pathogenic by multiple in silico prediction tools according to ACMG guidelines. It has not been reported in the large population frequency database gnomAD. This variant results in the substitution of a rigid proline with a polar histidine at position 59 within the N-terminal transactivation domain of the MAF protein. To date, this specific variant has been reported in three additional patients.1,10 Common manifestations among all four cases include midface hypoplasia, long philtrum, thin upper lip, and intellectual disability, while a small mouth was observed uniquely in our case (Table 2). We also note that the incidence of pericarditis appears higher than the average rate. It remains unclear whether this clinical specificity is related to particular amino acid substitutions. Based on these findings, our patient was definitively diagnosed with AYGRPS.
Comparative clinical features of patients with the MAF p.Pro59His pathogenic variant.
NA: not assessed.
The MAF gene encodes the Maf protein. 6 The Maf family is a subgroup of basic leucine zipper (bZIP) transcription factors, sharing homology with the prototype member, the v-Maf oncoprotein. Based on structural and functional differences, Maf family members can be classified into two major categories: small Maf proteins (e.g. MafK, MafF, MafG, MafT, and MafS) lack a transcriptional activation domain and act as transcriptional repressors when forming homodimers, while large Maf proteins contain a conserved N-terminal domain associated with transcriptional activation, form homodimers through their leucine zipper domains, and bind to relatively long conserved DNA sequences known as Maf recognition elements. Maf family proteins regulate various cell- and tissue-specific gene expression levels and play a significant role in cellular differentiation during development. 1 In addition, MAF participates in lens differentiation, as evidenced by its expression in the lens placode and primary lens fibers. Mutations in MAF result in dominant forms of cataracts. 19 Notably, the p.Pro59His variant identified in our patient is located within this critical N-terminal transactivation domain of the large MAF protein. The substitution of a rigid proline with a polar histidine at this conserved position is predicted to alter local protein conformation and potentially disrupt transcriptional activation function (Figure 3). This molecular perturbation provides a plausible explanation for the severe multisystem developmental defects observed in our case and in other patients with AYGRPS, as the MAF transcription factor regulates key genes involved in ocular, auditory, and skeletal development. 1
Given the multisystem nature of the disorder, management requires a multidisciplinary approach tailored to the individual’s specific needs. The care team may include ophthalmologists, otolaryngologists, developmental and behavioral specialists, speech therapists, occupational therapists, physiotherapists, orthopedists, endocrinologists, cardiologists, and neurologists. Treatment is primarily symptomatic and supportive, including interventions such as hearing aids for hearing loss, cataract extraction to improve vision, and antiepileptic medications for seizure control, supplemented by speech and motor skills training. 20 In this case, the child received sodium valproate for epilepsy, which effectively controlled the seizures. Additional supportive measures included a semi-liquid diet, use of diapers and a corrective chair during the day, and in-home educational support from a special education school. This case underscores the importance of individualized management plans in ultra-rare disorders. Furthermore, the successful use of sodium valproate provides valuable insight for future clinical decision-making in similarly affected individuals.
In summary, this study reports a severe phenotypic expression of AYGRPS caused by the MAF p.Pro59His variant, which notably extends the known severity spectrum of the disorder by documenting extreme postnatal growth failure and profound developmental delay. Our findings reinforce that in children presenting with the core constellation of symptoms, including epilepsy, intellectual disability, skeletal deformities, sensorineural hearing loss, congenital cataracts (or profound sensory deficits), and characteristic facial features, AYGRPS should be included in the differential diagnosis, and definitive genetic testing for the MAF gene is essential. A confirmed molecular diagnosis not only ends the diagnostic odyssey but also enables anticipatory management. Currently, management of AYGRPS remains primarily supportive and symptomatic, aimed at addressing specific issues such as seizures, hearing/vision impairment, and skeletal abnormalities to optimize quality of life and functional outcomes.
Footnotes
Acknowledgment
We gratefully acknowledge the child and the guardians for their cooperation in this study. DeepSeek was used for language improvement.
Authors’ contributions
Jingting Xu, Xiao Song, and Wei Jia contributed to conceptualization, formal analysis, and drafting of the original manuscript. Chaochun Zou conceptualized and designed the study, reviewed and revised the manuscript, and acquired funding. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Consent for publication
Informed consent for publication of clinical details and clinical images was obtained from the patient’s guardians.
Data availability
The datasets generated and analyzed during the current study are available in the ClinVar repository under accession number SUB15378131.
Declaration of conflicting interests
The authors declare no competing interests.
Ethics approval and consent for participate
The study was approved by the Ethical Committee of the Children’s Hospital of Zhejiang University School of Medicine. The Hangzhou Children's Social Welfare Center provided informed consent for the use of clinical information and genetic testing in this study. Informed consent to participate was obtained from the legal guardians.
Funding
This study was supported by the National Health Commission Scientific Research Fund (WKJ-ZJ-2409), the Clinical Innovation Team for Children with Hyperlipidemia (CXTD202501031), and the Children’s Hospital of Zhejiang University School of Medicine Pre-Research Fund (CHZJU2023YY001).