
Abstract
Pseudohypoparathyroidism is a rare genetic disorder characterized by hypocalcemia, hyperphosphatemia, and elevated serum intact parathyroid hormone levels. The taxonomy of pseudohypoparathyroidism is intricate, with heterogeneous clinical presentations and lack of specificity. A school-age Chinese girl was admitted with a 2-day history of fever and a single episode of seizure, with no clinical features of Albright hereditary osteodystrophy. Physical examination revealed positive Chvostek’s and Trousseau’s signs, with no other significant abnormalities. Laboratory tests revealed hypocalcemia, elevated intact parathyroid hormone levels, and transient hypokalemia (2.8 mmol/L). Whole-exome sequencing showed a 2.1-Kb deletion of exons 4–6 in syntaxin 16 (STX16); however, her parents did not exhibit any such alterations. The patient was finally diagnosed with pseudohypoparathyroidism and administered calcium gluconate perfusion, calcitriol, and elemental calcium. After discharge, her calcitriol and elemental calcium dosages were gradually tapered in the outpatient setting. By the 1-year follow-up, hypocalcemia had been largely corrected; however, her intact parathyroid hormone level remained above the normal reference range. This case of sporadic pseudohypoparathyroidism type Ib with a 2.1-Kb STX16 deletion underscores that pseudohypoparathyroidism can present as fever-induced seizure with transient hypokalemia, highlighting the importance of metabolic and genetic screening in children who experience seizures.
Introduction
Pseudohypoparathyroidism (PHP) is a rare genetic disorder characterized by the resistance of target cells to parathyroid hormone (PTH), leading to hypocalcemia, hyperphosphatemia, and significantly elevated serum levels of full-length parathyroid hormone (typically measured as intact PTH in clinical practice).1,2 The prevalence of PHP has been reported as 0.34 per 100,000 in Japan; however, its actual prevalence remains unknown. The main mechanisms involved are sporadic or autosomal dominant inherited genetic mutations and/or epigenetic alterations involving the guanine nucleotide-binding protein alpha-stimulating gene locus on chromosome 20q13. 3. 3
Several types of PHP exist, with PHP type Ia (PHPIa) and PHP type Ib (PHPIb) being the most prevalent. PHP presents with heterogeneous clinical manifestations and is prone to diagnostic oversights or inaccuracies. Patients with PHPIa frequently display characteristics of Albright hereditary osteodystrophy (AHO), including features such as round face, premature epiphyseal fusion, reduced height, shortened metacarpals and metatarsals, adiposity, neurocognitive impairments, and various ambiguous anomalies, whereas features of AHO are relatively uncommon in patients with PHPIb.
PHPIb, which is caused by maternal microdeletions within the GNAS locus or in the syntaxin-16 gene (STX16), falls under the category of inactivating PTH/PTH-related protein (PTHrP) signaling disorders (iPPSDs) type 3, according to the recent EuroPHP Network consensus, which classifies this condition as an epigenetic defect affecting the GNAS locus.4,5 STX16 harbors a cis-acting imprinting control region that maintains the methylation and imprinting status of the adjacent GNAS gene by interacting with transcription factors or other regulatory elements. In approximately 15%–20% of cases, the disorder is inherited in an autosomal dominant manner, primarily due to 3-kb deletions within the maternal allele of STX16. 6 Notably, 2.1-kb deletion encompassing exons 4–6 of STX16 has rarely been reported. 7
Herein, we present an unusual case of sporadic PHP combined with fever and hypokalemia in a Chinese female patient who had previously been diagnosed with febrile seizures.
Case report
A school-aged Chinese girl was admitted in December 2024 due to a 2-day history of fever and a single episode of generalized tonic-clonic seizure. The seizure, characterized by trismus, limb rigidity, and carpopedal spasm (“obstetrician’s hand” posture), lasted approximately 3 min and resolved spontaneously. Post-ictally, she was conscious with flushed skin. She had a history of two febrile seizures at ages 1 and 6 years, with previous extensive evaluations ruling out intracranial infection and epilepsy.
On admission, she was febrile (38.6°C) with stable vital signs. Her height was 130 cm (−1 to +1 SD), and weight was 29 kg (+1 to +2 SD). Neurological examination was unremarkable except for positive Chvostek’s and Trousseau’s signs, indicating latent tetany. No physical stigmata of AHO were observed. The physical examination of other systems was unremarkable.
Laboratory investigations revealed severe hypocalcemia (ionized calcium, 0.85 mmol/L) and transient hypokalemia (2.8 mmol/L). Her serum intact parathyroid hormone (iPTH) level was markedly elevated (398 pg/mL), consistent with parathyroid hormone resistance. Thyroid function, renal and hepatic parameters, and inflammatory marker levels were normal. Imaging studies, including thyroid/parathyroid ultrasonography, renal ultrasound, and brain computed tomography, showed no abnormalities. A nasopharyngeal swab was positive for influenza B virus.
Based on the biochemical profile of hypocalcemia, hyperphosphatemia, and elevated iPTH level, a diagnosis of PHP was established. To identify the genetic etiology, whole-exome sequencing was performed, which identified a heterozygous 2.1-kb deletion spanning exons 4–6 of the STX16 gene (Chr20: g.57244296_57246403), confirming PHPIb (Figure 1). Parental genetic testing did not detect this deletion, suggesting a de novo variant. Further methylation analysis was declined by the family.
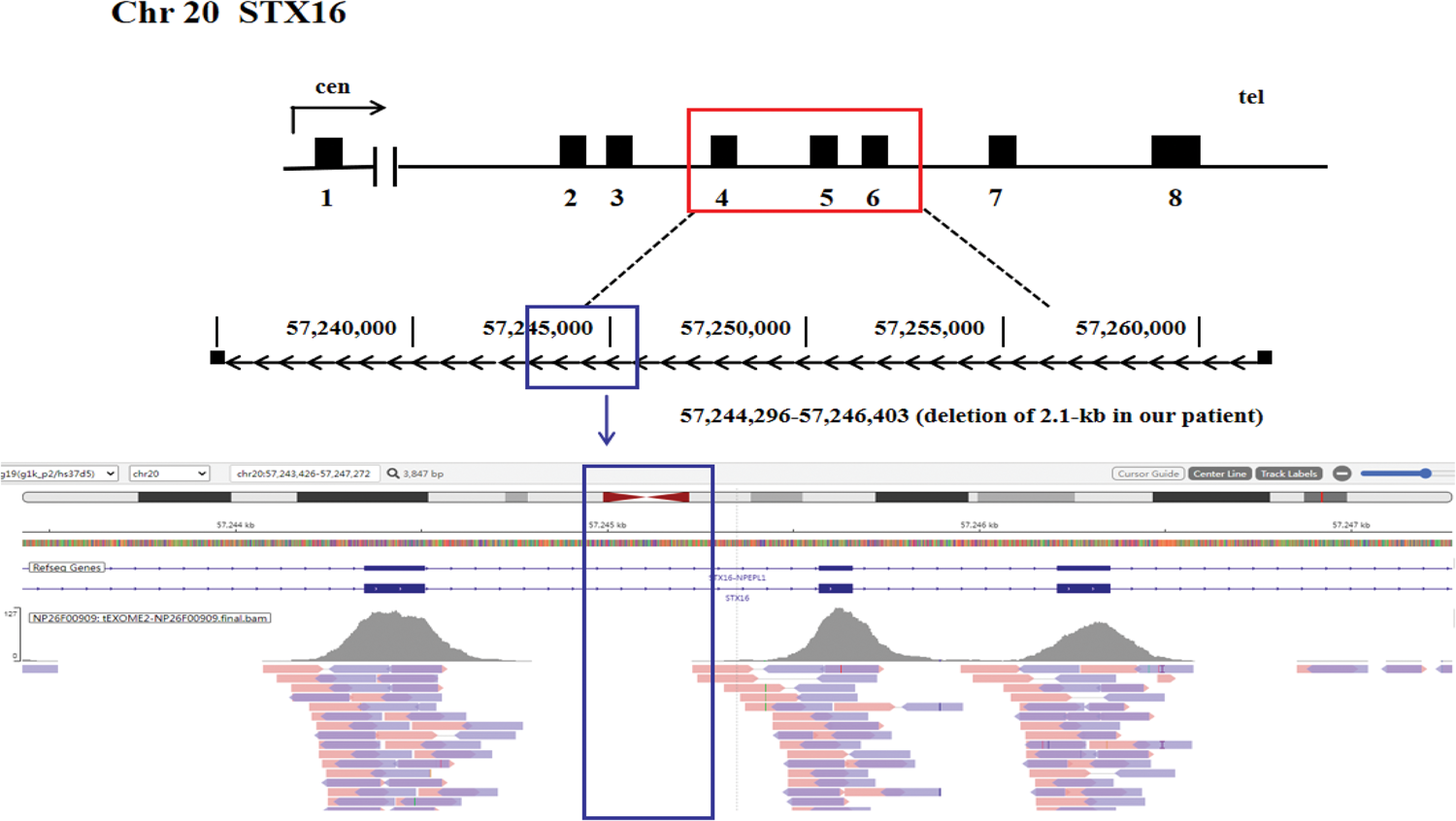
Results of whole exome capture analysis. Schematic representation of the heterozygous loss of exons 4–6 of syntaxin-16 (STX16) in the proband. Exons are indicated by black boxes; introns by lines; arrows show the direction of transcription. cen: centromeric; tel: telomeric. STX16 deletions are indicated in red boxes. The blue box represents a deletion of 2.1 kb from 57244296_57246403. The figure is not to scale.
Initial management included the administration of intravenous calcium gluconate followed by long-term oral calcitriol (0.015 μg/kg/day) and elemental calcium (15 mg/kg/day). The hypocalcemia and hypokalemia were promptly corrected, and no further seizures occurred. Concurrent antiviral therapy for influenza B led to fever resolution. During the 1-year follow-up, she was administrated oral calcitriol (0.005 μg/kg/day) and elemental calcium (5 mg/kg/day), which were then gradually tapered off. The patient has remained seizure-free with maintained normocalcemia and no adverse treatment effects.
All patient details have been deidentified, and written informed consent for publication was obtained from the patient’s parents. The Case Report (CARE) guidelines were strictly followed during the preparation of this case report. 8
Discussion
This report presents a case of PHPIb in a school-aged girl, manifesting with fever and seizure. The diagnosis was established based on characteristic biochemical abnormalities, including severe hypocalcemia, hyperphosphatemia, and elevated iPTH level, and was ultimately confirmed by the identification of a heterozygous 2.1-kb deletion in the STX16 gene. Moreover, the patient exhibited transient hypokalemia. This case highlights a critical clinical challenge; PHP represents a rare yet significant metabolic cause of seizure. Insufficient clinical awareness often leads to misdiagnosis and subsequent delay in appropriate management.
Our patient initially presented with fever and seizure and had been previously diagnosed with febrile seizures at ages 1 and 6 years. The primary complaint in our patient, who was >6 years old at the time of presentation, was fever accompanied with seizure. Although the differential diagnosis included fever-induced seizure plus (FS+), given the absence of a family history of FS+, febrile seizures, or epilepsy and owing to normal cranial imaging results, normal interictal electroencephalograph, and intact neurocognitive development, FS+ seemed unlikely. However, it is crucial to emphasize that FS+ is fundamentally an exclusive and retrospective diagnosis, whereas this patient exhibited a clear metabolic cause (hypocalcemia) that can directly precipitate seizures. Although FS+ is associated with several genes (e.g. SCN1A, SCN1B, and GABRG2) and exhibits genetic heterogeneity (with some cases lacking detectable mutations), genetic testing in this patient did not reveal any of the aforementioned pathogenic variants. More importantly, following targeted calcium supplementation, the patient remained seizure‑free during the 1‑year follow‑up period, with sustained normal neurodevelopment. This clinical trajectory does not support a diagnosis of FS+. Nonetheless, ongoing close monitoring of the patient’s neurodevelopmental progress and long‑term follow‑up remain essential for final diagnostic confirmation.
Additionally, the patient experienced fever for 2 days before the seizure that lasted 3 min and resolved spontaneously. There was no status epilepticus, and the patient’s consciousness was normal after the seizure. Throughout the illness, there were no signs of abdominal pain, rash, or joint pain. Blood inflammatory markers, EEG, and cranial imaging findings were normal. There was no significant family history, and follow-up did not indicate any neurological deficits. Moreover, whole-exome sequencing (WES) did not reveal mutations in related genes such as MEFV, SCN1A, and PCDH19.9–11 Previous episodes excluded the possibility of intracranial infections, autoimmune encephalitis, neoplastic diseases, and metabolic disorders. Therefore, familial Mediterranean fever, febrile infection-related epilepsy syndrome, and Dravet Syndrome were considered unlikely.
Furthermore, hypokalemia is an uncommon and typically transient manifestation in patients with PHPIb. In this case, the patient exhibited a transient decline in serum potassium levels; however, the underlying etiology was not attributable to renal potassium loss (as evidenced by decreased urinary potassium excretion) or extrarenal losses (e.g. via vomiting or diarrhea). The patient’s blood pressure remained normal; there was no evidence of metabolic alkalosis, dietary potassium intake was adequate, thyroid function was within normal limits, and there was no history of medication use (such as insulin) that could induce intracellular potassium shifts. Moreover, there was no family history of periodic paralysis. Notably, the hypokalemia resolved concurrently with the normalization of serum calcium and iPTH levels, suggesting an association between the two abnormalities.
12
The causes of PHPIb combined with hypokalemia were as follows:
It may be secondary to the dysregulation of calcium/PTH/vitamin D caused by PHP. Low vitamin D3 levels can lead to high transcriptional activation of the renin–angiotensin–aldosterone system via vitamin D receptors, thereby reducing serum potassium levels.13,14 The G protein stimulatory alpha subunit (Gsα)/cyclic adenosine monophosphate (cAMP)/protein kinase A signaling pathway enhances potassium channel activity. In patients with PHP, renal resistance to PTH inhibits this pathway, leading to potassium recirculation disorders.
12
Moreover, hypokalemia as well as high renin and high aldosterone levels are characteristic features of Gitelman syndrome and Bartter syndrome. The patient exhibited normal growth and development, with no evidence of hypomagnesemia, hypochloremia, or metabolic alkalosis. Additionally, the urinary potassium and chloride levels were within normal ranges, thereby reducing the likelihood of Gitelman and Bartter syndromes. WES results further supported the exclusion of both syndromes. Moreover, the transient hypokalemia may be associated with fever itself rather than being caused by PHPIb.
Deletion of exons 4–6 in the STX16 gene represents a classic pathogenic mutation responsible for autosomal-dominant sporadic PHPIb. This deletion has been consistently identified as the most prevalent molecular defect across multiple studies. In the present case, the deletion was not detected in either parent, strongly suggesting a de novo event. Although a maternally inherited 3‑kb deletion within STX16 is widely recognized as the primary cause of autosomal dominant (AD)‑PHPIb, this inheritance pattern does not appear to apply to sporadic cases such as ours. In fact, recent investigations have demonstrated that approximately 5%–10% of sporadic PHPIb patients harbor de novo STX16 deletions, 15 and additional reports of sporadic cases carrying the 3‑kb STX16 deletion have been documented. 16 Two principal hypotheses have been proposed to elucidate the molecular mechanisms by which this deletion impairs PTH function, potentially acting independently or synergistically. First, the deletion may disrupt a cis‑acting imprinting control element located upstream of the GNAS locus, leading to aberrant methylation and transcriptional silencing of the maternal allele. This epigenetic alteration results in reduced expression of the Gsα protein, thereby compromising calcium reabsorption in the distal renal tubules. 17 Second, the deletion may interfere with long‑range chromatin interactions between the STX16 imprinting control region and other regulatory elements within the GNAS imprinted region, such as the NESP‑imprinting control regio (ICR). 18 Both mechanisms ultimately culminate in renal resistance to PTH. Given the potential for asymptomatic carriage across multiple generations and the sporadic nature of the disease, our findings underscore the importance of comprehensive genetic and methylation testing for all patients with clinical and biochemical suspicion of PHPIb, irrespective of family history. Such testing is pivotal for establishing an accurate diagnosis, providing informed genetic counseling, and facilitating effective family management.
The limitation of our study was the inability to perform comprehensive molecular profiling, including assessment of urinary cAMP, Gsα activity, and GNAS methylation status. Despite the lack of the above analysis, the diagnosis of sporadic PHPIb was supported by the absence of AHO features, characteristic biochemical findings, positive responses to calcitriol and calcium supplementation, and identification of an STX16 exon deletion. Our findings align with previously published cases such as that reported by Ramalho et al. 16
Overall, we presented a sporadic case of PHPIb that manifested with fever‑induced seizure and transient hypokalemia. Genetic analysis identified a heterozygous 2.1‑kb deletion encompassing exons 4–6 of the STX16 gene, which confirms the diagnosis and underscores the utility of molecular testing in clarifying ambiguous presentations. This report highlights that PHP should be considered in the differential diagnosis of seizure, especially when accompanied with electrolyte disturbances. The diagnostic approach and longitudinal follow-up of the present case have yielded valuable insights that can inform clinical decision-making and guide the implementation of appropriate treatment strategies.
Footnotes
Acknowledgments
None.
Author contributions
Writing-original draft preparation: XM and JR
Writing-review and editing: CQ and SY
Data curation: YH
Supervision: CQ and SY
Project administration: XM, JR, CQ, and SY
Data availability statement
Data analyzed in this study were obtained from publicly available sources.
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Ethics statement
Written informed consent for the publication of the details and images of the patient was obtained from her legal representatives. Exemption from the requirement for ethics approval for the present study was granted by the Ethics Committee of The First Affiliated Hospital of Chengdu Medical College (Approval No. 2025CYFYIRB-BA-007, approved in January 2025). We also declare that this manuscript has not been shared as a preprint.
Funding
This work was supported by the High-level Talent Introduction Project of the First Affiliated Hospital of Chengdu Medical College (Grant no. CYFY-GQ64, CYFY-GQ69) and Scientific Research Project of Chengdu Medical Association (No: 2024155).