
Abstract
Anomalous origin of the left coronary artery from the pulmonary artery is a rare congenital coronary anomaly. This case report describes an adult male with anomalous origin of the left coronary artery from the pulmonary artery, in whom transthoracic echocardiography revealed mitral valve prolapse without apparent anomalies of the coronary ostia. The definitive anatomical diagnosis was established using invasive coronary angiography and coronary computed tomography angiography. This case underscores the indispensable value of multimodality imaging in the diagnosis of anomalous origin of the left coronary artery from the pulmonary artery.
Keywords
Introduction
Anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA) is a rare type of congenital heart disease that usually presents with symptoms of heart failure at approximately 3 months of age. Without early surgical correction, the majority of untreated patients die before 1 year of age. This may account for the extreme rarity of adult-type ALCAPA. Some patients can remain asymptomatic for decades because of well-developed coronary collateral circulation. 1 Echocardiography typically serves as the initial diagnostic tool, whereas advanced modalities such as invasive coronary angiography (ICA), coronary computed tomography angiography (CCTA), and cardiac magnetic resonance (CMR) are pivotal for definitive diagnosis and comprehensive prognostic assessment. 2 We report the case of ALCAPA in an older patient to highlight the diagnostic challenges and management strategies in late adulthood.
Case presentation
This study complies with the Case Report (CARE) guidelines. 3
In August 2025, a male in his 60s was admitted to the Department of Cardiology at Gansu Province People’s Hospital in Lanzhou, China. He presented with a 3-year history of intermittent exertional chest tightness and shortness of breath, which had worsened and was associated with dizziness for 3 days. He had no significant past medical, personal, systemic, or family history. Physical examination revealed a blood pressure of 148/102 mmHg (1 mmHg =0.133 kPa), a respiratory rate of 19 breaths per min, and a heart rate of 65 beats per min. There was no precordial bulge. A grade 3/6 continuous systolic murmur was audible over the second and third intercostal spaces along the left sternal border.
Laboratory tests showed elevated high-sensitivity troponin I (0.0368 ng/mL; reference value <0.0262 ng/mL) and elevated N-terminal pro-B-type natriuretic peptide (616.40 pg/mL; reference value <125 pg/mL). Electrocardiography indicated sinus rhythm, intraventricular conduction delay, and ST-segment depression of 0.1 mV in leads V4–V6 (Figure 1). Echocardiography demonstrated prolapse of the A2 segment of the mitral valve accompanied by mild-to-moderate regurgitation, left atrial enlargement, left ventricular wall hypertrophy, and preserved left ventricular ejection fraction. The ostium of the left coronary artery appeared normal (Figure 2). The patient was diagnosed with acute non-ST-segment elevation myocardial infarction.
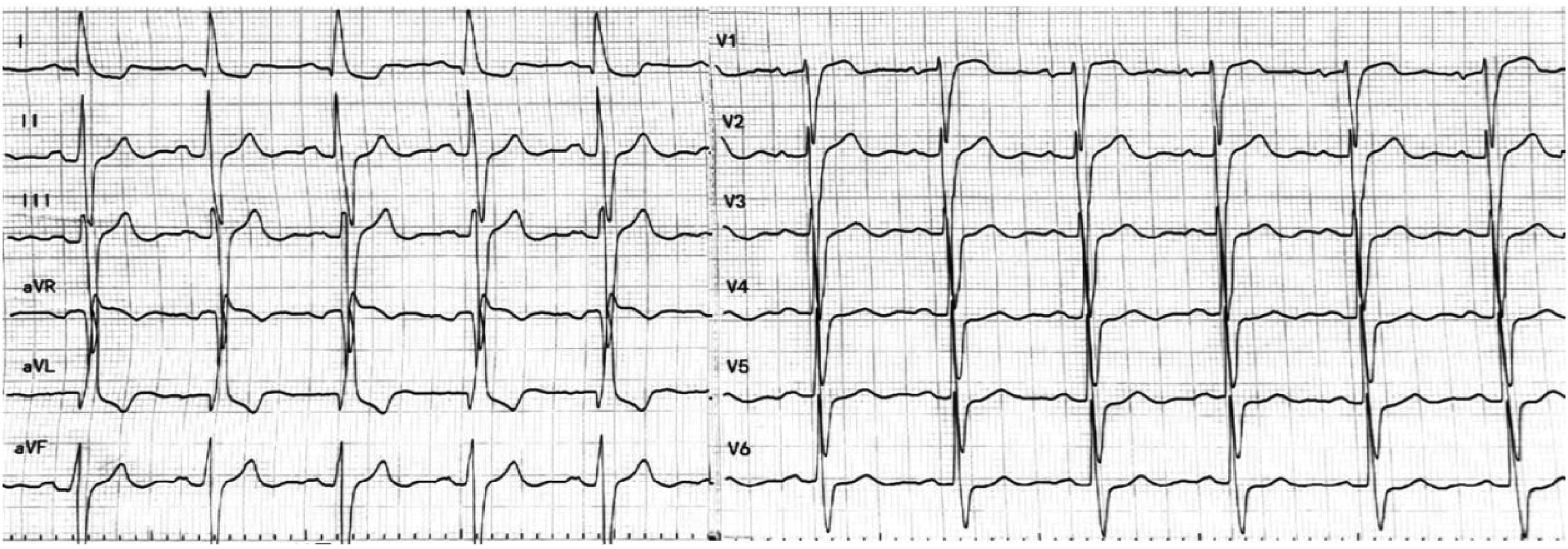
The ECG revealed sinus rhythm with nonspecific intraventricular conduction delay and ST-segment depression (0.1 mV) in leads V4–V6. ECG: electrocardiogram.

Echocardiography demonstrated normal origins of both coronary arteries but revealed mitral valve prolapse with associated regurgitation. (a) Left coronary artery ostium; (b) right coronary artery ostium; (c) prolapsing mitral valve leaflet; and (d) regurgitant jet on color Doppler.
However, CCTA revealed that the left coronary artery anomalously originated from the right aspect of the pulmonary trunk and followed an interarterial course between the aorta and pulmonary artery. The ostium at the left coronary cusp was, in fact, a blind-ending stump. The right coronary artery was tortuous and dilated, reflecting a compensatory adaptation. Abundant collateral vessels were observed supplying the territories of the posterior descending artery, the left anterior descending artery, and the interventricular septum (Figure 3).

(a, b) The left coronary artery (arrow) anomalously originates from the right aspect of the main pulmonary artery (asterisk). (c) The right coronary artery is markedly tortuous and dilated, with abundant collateral vessels visualized.
ICA confirmed a right-dominant coronary system. The right coronary artery was dilated throughout its course, and its distal segments perfused the left coronary system retrogradely through collateral vessels (Figure 4).

Coronary angiography was notable for a significantly dilated and tortuous right coronary artery, which provides robust retrograde collateral flow to the left coronary territory.
Based on a comprehensive multimodality imaging assessment, the final diagnosis of ALCAPA was confirmed. Considering the institutional resources and the patient’s clinical status, we proposed two management options: coronary artery bypass grafting (CABG) with ligation of the left coronary artery or conservative medical therapy. After a thorough discussion of the potential risks and outcomes of both interventions, the patient opted for medical management over corrective surgery. Therefore, a medical management strategy was adopted, and the patient was discharged on a regimen of bisoprolol extended release 5 mg q.d. At the 1-month telephone follow-up, the patient reported strict adherence to the prescribed medication regimen and marked improvement in symptoms. Furthermore, no complications or adverse events were observed during this period. A 6-month follow-up was planned, including echocardiography, electrocardiogram (ECG), and other relevant assessments.
Discussion
ALCAPA is a rare congenital cardiac malformation with high lethality if left untreated. Although its reported prevalence is approximately 1 in 300,000, the true incidence is likely higher, as some cases remain undiagnosed due to the absence of typical clinical symptoms. 2 Adult-type ALCAPA is even rarer and exhibits a broad spectrum of clinical manifestations. The symptom profile largely depends on two pathophysiological factors: the degree of collateral circulation and the severity of coronary steal syndrome. To compensate, the dominant right coronary artery undergoes significant dilatation, often resulting in a right-dominant coronary distribution with abundant collateral vessels aimed at preserving myocardial perfusion. 4 However, this compensatory mechanism triggers a vicious cycle of “ischemia-steal-worsening ischemia,” which forms the core pathological basis for myocardial ischemia and sudden cardiac death in patients with ALCAPA.
The diagnostic challenge in our patient is similar to that of a previously documented case, in which a symptomatic adult female presented with angina and palpitations that were repeatedly missed on successive echocardiogram. 5 These pitfalls underscore the necessity of multimodality imaging in ALCAPA. Echocardiography, as a noninvasive and safe modality, allows evaluation of cardiac structure, function, coronary artery ostia, and flow direction. Unexplained mitral valve prolapse, even when associated with mild regurgitation, should prompt consideration of ALCAPA. 6 It currently serves as a key tool for the initial diagnosis and follow-up of ALCAPA. 7 It is recommended to evaluate anomalous coronary arteries using coronary angiography either through catheterization, computed tomography, or CMR. ICA was the standard for diagnosing ALCAPA, but it has been replaced by noninvasive methods. CCTA has gradually become the gold-standard technique for confirming the definitive diagnosis, providing direct visualization of the vascular anatomy through three-dimensional reconstruction of the coronary tree. Meanwhile, it plays a crucial role in the formulation of surgical treatment plans. CMR is useful for detecting the presence of myocardial scar through late gadolinium enhancement (LGE) in older children and adults, providing important information for assessing the risk of sudden cardiac death (SCD) and the necessity of an ICD. 8 Furthermore, the risk of SCD increases substantially when the anomalous coronary artery passes between the aorta and pulmonary artery or demonstrates ostial stenosis. 9 Because the patient was unable to tolerate imaging due to claustrophobia, we could not assess the risk of SCD. International guidelines recommend surgical correction for ALCAPA, regardless of age or symptom status, to restore a dual-coronary artery system. 10 Common surgical approaches include direct coronary artery reimplantation, transpulmonary baffling (Takeuchi procedure), and CABG. However, in asymptomatic older patients with moderate chronic ischemia, survival with medical management may be feasible. 2 The choice of procedure depends on the anatomy and experience of the center, emphasizing the importance of individualized assessment to determine the most appropriate treatment strategy.
Conclusion
Adult-type ALCAPA is an exceedingly rare yet clinically significant congenital anomaly. The synergy of a multimodality imaging approach is indispensable for achieving accurate diagnosis and providing comprehensive evidence for clinical decision-making.
Footnotes
Acknowledgments
The authors used ChatGPT in order to improve language. After using the tool, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication. We are grateful to the patient and his family for providing informed consent for publication of the case.
Author contributions
Manuscript preparation: Li Xinghui and Wang Xin. Data acquisition: Zhang Xiaoming and Qin Yahong. Manuscript revision: Wei Rong. All of the authors have read and approved the final manuscript.
Data availability statement
Data will be made available on request.
Disclosures
The authors declared no potential conflicts of interest with respect to the research, authorship, and publication of this article.
Ethic statement
The study was approved by the Ethics Review Committee of Gansu Province People’s Hospital (approval number 2025-661), Lanzhou, China, in December 2025. All identifiable patient information has been removed. Signed informed consent for treatment and publication was obtained from the patient of this case report in September 2025.
Funding
This work was supported by the Natural Science Foundation of Gansu Province (Grant No. 25JRRA312).