
Abstract
Hypoparathyroidism is a rare endocrine condition characterized by insufficient secretion of parathyroid hormone (PTH), resulting in abnormally low calcium levels (hypocalcemia) and elevated phosphate levels (hyperphosphatemia) in the blood. This report describes a man in his late 30s with a chronic skin condition marked by dryness and desquamation. He occasionally experienced mild perioral numbness. Over the past year, he developed recurrent neuromuscular irritability, including worsening perioral numbness, tingling or numbness in the hands and feet, and muscle spasms consistent with tetany. He was diagnosed with hypoparathyroidism, and his symptoms improved markedly after calcium and calcitriol supplementation. Genetic testing revealed a novel heterozygous c.2298C>G (p. Tyr766Ter) mutation in exon 18 of the fibroblast growth factor receptor 1 gene. This case report aimed to describe this novel mutation and its potential role in the pathogenesis of primary hypoparathyroidism and to discuss relevant diagnostic and therapeutic management strategies. In addition, it broadens our understanding of genetic mutations associated with hypoparathyroidism and provides clinically relevant diagnostic information that may benefit future patients with the similar genetic alteration. Furthermore, it underscores the importance of genetic analysis in elucidating the heterogeneity and complexity of hypoparathyroidism, thereby supporting the development of more precise and tailored treatment approaches.
Keywords
Introduction
Hypoparathyroidism (HypoPT) is a rare endocrine disorder characterized by inadequate secretion of parathyroid hormone (PTH), resulting in reduced calcium levels (hypocalcemia) and increased phosphate levels (hyperphosphatemia) in the bloodstream. 1 The estimated prevalence of HypoPT is approximately 37 per 100,000 individuals in the United States 2 and 22 per 100,000 in Denmark, 3 with lower prevalence reported in other countries.4,5 HypoPT presents with a range of acute and chronic clinical manifestations. Acute hypocalcemia can cause cardiac or neuromuscular irritability, clinically presenting as impaired cardiac contractility, arrhythmias, and sensory nerve and muscle dysfunction. Chronic hypocalcemia may lead to basal ganglia calcifications (Fahr syndrome), posterior subcapsular cataracts, cardiac failure, increased bone mineral density, and various dermatological manifestations. Calcium and vitamin D supplementation for HypoPT may be associated with kidney-related complications, such as kidney stones and renal insufficiency.2,6 The most common cause of HypoPT is often the inadvertent removal or damage of the parathyroid glands during neck surgery, accounting for approximately 75% of cases. 7 The remaining approximately 25% cases result from a variety of factors, including autoimmune disorders, magnesium deficiency, genetic conditions, and other rare causes. 8 Although genetic causes account for fewer than 10% of all HypoPT cases, 9 molecular analyses have identified an increasing number of genes that, when defective, result in impaired formation of the parathyroid glands, disordered synthesis or secretion of PTH, or postnatal destruction of the parathyroid glands.9,10
To the best of our knowledge, we report the first case of HypoPT associated with c.2298C>G mutation in the fibroblast growth factor receptor 1 (FGFR1) gene in a Chinese man along with his clinical and genetic findings.
Case description
Clinical characteristics
The patient was a Chinese man in his late 30s with a long-standing history of chronic dry skin, desquamation, and perioral numbness. These symptoms were congenital, with no identifiable triggers. He could not recall the precise onset of these symptoms. The symptoms had been relatively mild, and he had not sought medical attention. There was no family history of related disorders. He is married with two children, and among his immediate relatives, including his children, there were no reported cases of hypocalcemia, early cataracts, seizures, anosmia, or hypogonadism. Over the past year, he experienced recurrent neuromuscular irritability, manifested as worsening perioral numbness, numbness or tingling in the hands and feet, and tetany. These episodes occurred approximately once a week and were not preceded by any identifiable trigger. In March 2024, he presented to the emergency department of The Fourth Affiliated Hospital of Soochow University with severe numbness around his mouth and extremities. He arrived at the emergency room walking by himself. His vital signs on presentation were as follows: blood pressure, 154/96 mmHg; pulse, 85 beats/min; regular and full in volume, respiratory rate, 20 breaths/min; and temperature, 36.5°C. Continuous cardiac monitoring revealed no significant electrocardiographic abnormalities. Physical examination findings of other systems were unremarkable. The patient had no history of neck surgery, radiation exposure, or autoimmune diseases.
Laboratory tests revealed a serum calcium level of 1.41 mmol/L (normal range, 2.11–2.52 mmol/L), consistent with hypocalcemia, and a serum phosphorus level of 1.64 mmol/L (normal range, 0.85–1.51 mmol/L), indicative of hyperphosphatemia. Serum potassium level was slightly reduced at 3.46 mmol/L (reference range, 3.5–5.3 mmol/L). The patient received an intravenous injection of calcium gluconate and an infusion of potassium chloride, which gradually relieved the numbness. To identify the cause of hypocalcemia, he was evaluated in the endocrinology department the following day. Testing showed a suppressed PTH level of 6.6 pg/mL (normal range, 15–65 pg/mL) and a serum magnesium level of 0.94 mmol/L (normal range, 0.75–1.02 mmol/L). Bone turnover markers indicated reduced bone formation, with osteocalcin decreased to 5.27 ng/mL (normal range, 14–42 ng/mL), and reduced bone resorption, with β-CrossLaps (CTX) reduced to 0.06 ng/mL (normal range, 0.225–0.936 ng/mL). Other laboratory tests, including hemoglobin A1c (HbA1c), thyroid function, creatinine, beta-2 microglobulin, 25-hydroxyvitamin D, pituitary hormones, insulin-like growth factor-1, and testosterone, were within normal limits. Ultrasound findings revealed no abnormalities in the parathyroid glands. Bone mineral density assessment performed with dual-energy X-ray absorptiometry showed normal results, with Z-scores of −1.1 at the femur neck and −1.5 at the lumbar spine. Following the diagnosis of HypoPT, no other diagnosis was established. The patient provided written informed consent for treatment and was prescribed oral calcium carbonate tablets (0.75 g twice daily) and calcitriol (0.25 µg three times daily). During the course of treatment, he remained stable and asymptomatic. Following 45 days of treatment, reevaluation showed a PTH level of 7.93 pg/mL, calcium of 2.07 mmol/L, and normal phosphorus and magnesium levels of 1.5 mmol/L and 0.96 mmol/L, respectively. During a telephone follow-up, the patient reported adherence to the prescribed oral calcitriol and calcium supplements, with no change in the dosage and no adverse and unanticipated events.
Genetic results
Peripheral blood samples were collected in ethylenediaminetetraacetic acid (EDTA) tubes from the proband for whole-genome extraction. His parents, sisters, and children declined genetic testing, as they did not exhibit any apparent symptoms. Whole-exome sequencing, performed using next-generation sequencing and Sanger sequencing, identified a novel heterozygous variant in exon 18 of the FGFR1 gene, denoted as c.2298C>G (p. Tyr766Ter) (Figure 1). In the next-generation sequencing analysis, the variant allele frequency was 47%, with a total of 308 reads, including 164 wild-type reads and 144 mutant reads. According to the American College of Medical Genetics and Genomics (ACMG) guidelines, this FGFR1 mutation was classified as of “uncertain significance,” based on criteria PM2 (supporting: the variant is absent or extremely rare in population databases) and PVS1 (moderate: the mutation is predicted to cause impaired protein function). The FGFR1 gene sequence (NM_023110.3) was obtained from the National Center for Biotechnology Information (NCBI) database (https://www.ncbi.nlm.nih.gov/). Furthermore, three-dimensional models of the wild-type and mutant FGFR1 proteins (NM_023110.3: c.2298C>G) were generated using the SWISS-MODEL online server. The global model quality estimation (GMQE) score for the mutant FGFR1 was 0.78, with 100% sequence identity, indicating high-quality model predictions. These models are essential for evaluating the potential structural and functional impacts of the identified mutation. The c.2298C>G (p. Tyr766Ter) mutation is a nonsense variant that causes premature termination of the protein at tyrosine 766, resulting in the truncation of 56 amino acids from the wild-type protein, which normally comprises 822 amino acids (Figure 2).

Gene sequencing of the patient revealed a novel heterozygous nonsense mutation in FGFR1, c.2298C>G (exon18). (a) Next-generation sequencing; the sites marked with red dotted lines indicate the mutation sites and (b and c) Sanger sequencing of sense and antisense strands, respectively; the red arrow indicate the mutation sites. FGFR1: fibroblast growth factor receptor 1.

Difference between the wild-type and mutant FGFR1 protein (p. Tyr766Ter) (regional model). The left model represents the wild-type protein, and the right model represents the mutant. Blue covered part indicates the 56 amino acids truncated in the mutant protein. FGFR1: fibroblast growth factor receptor 1.
Discussion and conclusion
Although genetic disorders rarely constitute the primary cause of HypoPT, identifying underlying genetic factors is essential for effective management. Accurate genetic diagnosis facilitates the development of targeted therapies tailored to specific genetic anomalies, thereby improving treatment outcomes. 9 Based on molecular genetic analyses, researchers have identified over 15 pathogenic genes associated with HypoPT till date, 9 underscoring the importance of genetic research in understanding disease complexity and guiding therapeutic strategies. Specific genes, including GCM2, 11 SOX3, 12 and PTH 13 have been associated with isolated HypoPT. Mutations in these genes primarily affect the embryonic development of parathyroid glands, often leading to this form of the disease. Other genes, such as GATA3, 14 TBX1, 15 CHD7, 16 SEMA3E, 17 and TBCE, 18 are implicated in syndromic forms of HypoPT, where the condition occurs as part of broader disorders, including DiGeorge syndrome 19 and autoimmune polyendocrine syndrome type 1. 20 Additionally, HypoPT can coexist with other significant medical conditions such as renal disease and mitochondrial disorders.6,21 These associations highlight the need for a holistic diagnostic and management approach that considers all underlying conditions to optimize patient care.
Our study identified a novel FGFR1 variant in a Chinese male patient with HypoPT. The FGFR1 gene, located on chromosome 8p11.23, comprises 18 exons. 22 The FGFR1 protein encoded by this gene, functions as a receptor tyrosine kinase (RTK) that plays a critical role in normal cellular processes and pathological conditions, including cell growth, survival, migration, and apoptosis, underscoring its potential relevance for medical intervention. 23 Mutations in the FGFR1 gene are primarily associated with idiopathic hypogonadotropic hypogonadism (IHH), a rare endocrine disorder characterized by gonadal dysplasia; in some patients, this condition is accompanied by olfactory dysfunction known as Kallmann syndrome (KS).24–30 This patient exhibited normal sex hormone levels and gonadal function, allowing IHH to be ruled out. The ClinVar database contains records of 91 additional nonsense mutations within the tyrosine kinase domain (TKD) of FGFR1 (https://www.ncbi.nlm.nih.gov/clinvar/) (Figure 3); however, none have been reported at amino acid position 766. Further investigation is warranted to determine whether this specific mutation within the TKD of FGFR1 affects PTH levels.
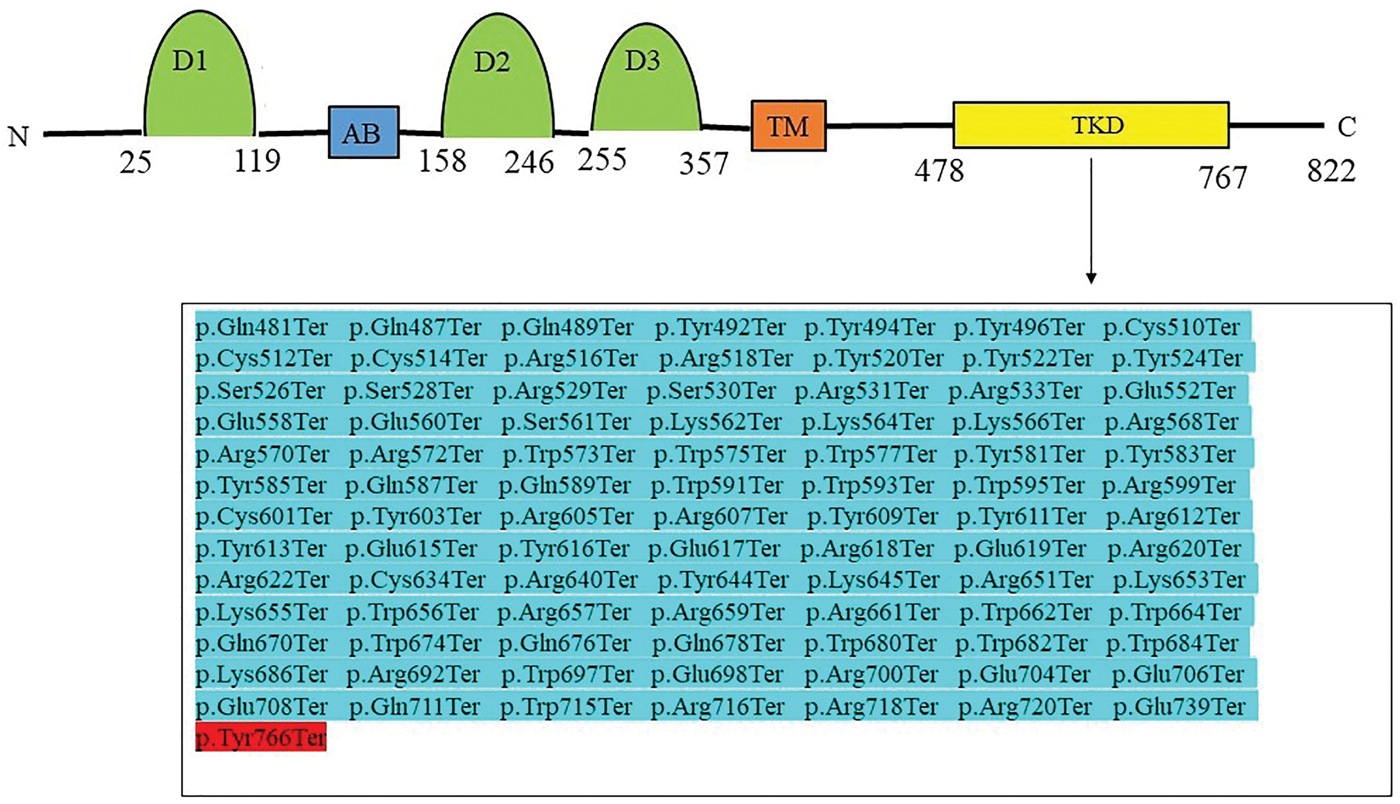
Variants reported in the TKD of FGFR1. The amino acids affected by the FGFR1 c.2298C>G mutation identified in this study are highlighted in red. AB: acidic box; TM: transmembrane; TKD: tyrosine kinase domain; FGFR1: fibroblast growth factor receptor. 1.
FGFR1 is expressed in multiple tissues and organs, including the kidneys, lungs, bones, and parathyroid glands.31–33 The FGFR1 protein consists of an extracellular region containing three immunoglobulin (Ig)-like domains (D1, D2, and D3) that confer ligand-binding affinity and specificity, a single hydrophobic transmembrane segment, and a cytoplasmic TKD with protein tyrosine kinase activity. 34 FGFR1 interacts with ligands, such as fibroblast growth factor (FGF) and heparan sulfate (HS), leading to receptor dimerization and activation of the receptor complex. This process initiates downstream signaling pathways through tyrosine kinase activity, which underpins essential physiological functions 35 including embryonic development, organ formation, tissue repair and remodeling, angiogenesis, and various metabolic activities.36–38 FGFR1 is also expressed in human skin tissues, and FGFs are involved in wound healing. 39 It remains unclear whether skin dryness is part of the broader phenotypic spectrum associated with FGFR1 mutation. However, the patient’s symptoms of dry skin and desquamation improved markedly following calcium supplementation. Within the parathyroid gland, FGF23 interacts with the klotho–FGFR1 receptor complex, leading to reduced PTH gene expression and decreased PTH secretion through activation of the mitogen-activated protein kinase (MAPK) pathway, as reported by studies conducted on rats and in vitro cultures of parathyroid tissue.31,40 Therefore, FGFR1 plays a significant role in the pathogenesis of parathyroid disease and the regulation of PTH expression. Specific mutations in the TKD are known to be pathogenic, primarily through nonsense-mediated mRNA decay and protein truncation.41,42 Internal tandem duplications (ITD) within the TKD of FGFR1 leads to constitutive receptor activation, enhancing cell growth, differentiation, and survival via Ras/MAPK signaling. 43 The FGFR1 mutation identified in our study results in the premature termination at tyrosine 766 within the TKD, truncating 56 amino acids. Sequence and structural analyses suggest that this truncation may impair FGFR1 function. We hypothesized that this truncation may result in sustained MAPK signaling, leading to reduced PTH expression and secretion.
There are several limitations to our study. The source of the patient’s FGFR1 mutation could not be determined because his family members declined genetic testing, making it difficult to determine the mutation’s origin and inheritance patterns. In addition, the absence of whole-genome sequencing prevents the identification of other potential genetic abnormalities in noncoding regions that may contribute to HypoPT. Furthermore, the absence of a pathological examination of the parathyroid gland makes it difficult to determine whether tissue or cellular damage, immune-mediated inflammation, or other factors contribute to HypoPT, in addition to FGFR1 gene mutations. To address these uncertainties, studies using animal models are required. Developing a knockout model carrying this mutation could provide critical insights into whether the genetic alteration directly causes HypoPT. Such studies are essential for establishing a definitive link between FGFR1 mutations and the development of HypoPT, improving our understanding of the disease etiology, and informing future therapeutic strategies for effective treatments.
In conclusion, this case report identified a novel FGFR1 nonsense variant in a Chinese man with HypoPT. Our findings expand the spectrum of mutations associated with HypoPT and provide important diagnostic insights for future patients with similar genetic alteration. Furthermore, this study highlights the value of comprehensive genetic screening and suggests avenues for targeted therapeutic interventions, thereby improving understanding of the genetic basis of HypoPT.
Footnotes
Acknowledgments
The authors would like to thank the proband and his family members for participating in and supporting this study. We would also like to thank the innovation transformation platform of The Fourth Affiliated Hospital of Soochow University.
Author contributions
HS designed the study; XC drafted and revised the manuscript; XC, YZ, and RS acquired, analyzed, and interpreted the data. HS critically reviewed the article. All authors have read and approved the final manuscript.
Consent for publication
Written informed consent was obtained from the individual for the publication of any potentially identifiable images or data included in this article. We have deidentified all patient details.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: MedGen UID: 289648, concept ID: C1563720.
Declaration of conflicting interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Declaration of reporting guidelines
The reporting of this study conforms to the Case Report (CARE) guidelines. 44
Ethics approval and consent to participate
This study received approval for publication from the Medical Ethics Committee of The Fourth Affiliated Hospital of Soochow University on 26 November 2025, with the approval number 251311.
Funding
This work was supported by the National Natural Science Foundation of China (grant no. 82270755 to HS), the Suzhou Science and Technology Project (grant no. SZM2022013 to HS), and the Gusu Talent Program (grant no. GSWS2023020 to HS).
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.