
Abstract
Primary hyperparathyroidism is characterized by hypercalcemia and hypophosphatemia caused by excessive parathyroid hormone secretion. Gitelman syndrome, a rare autosomal recessive salt-losing tubulopathy, presents with hypokalemia, hypomagnesemia, hypocalciuria, and secondary hyperaldosteronism. The coexistence of these conditions leads to an atypical clinical picture and may result in a refractory hypercalcemic crisis that responds poorly to standard therapies. We describe a case of refractory hypercalcemia accompanied by hypophosphatemia, elevated parathyroid hormone levels, hypokalemia, hypomagnesemia, and, notably, hypocalciuria that was inconsistent with the degree of hypercalcemia. The patient experienced recurrent hypercalcemic crises that required treatment with zoledronic acid and denosumab after conventional interventions failed. The diagnosis was ultimately confirmed through postoperative pathology and genetic testing. This case emphasizes that hypocalciuria in the setting of persistent hypercalcemia—especially when severe hypomagnesemia is present—should raise suspicion for underlying Gitelman syndrome. Early identification of this coexistence is essential, as optimal management requires surgical treatment of primary hyperparathyroidism followed by targeted correction of electrolyte abnormalities associated with Gitelman syndrome, ultimately improving clinical outcomes.
Introduction
Primary hyperparathyroidism (PHPT) is a common endocrine disorder characterized by hypercalcemia and hypophosphatemia resulting from excessive parathyroid hormone (PTH) secretion. It often presents with bone demineralization, renal stone formation, or neuromuscular complications and is typically caused by parathyroid adenomas, hyperplasia, or malignancy. In contrast, Gitelman syndrome (GS) is a rare autosomal recessive renal tubular disorder first described in 1966. 1 It is caused by loss-of-function mutations in the SLC12A3 gene, which encodes the thiazide-sensitive sodium–chloride cotransporter (NCC) located in the distal convoluted tubule. 2 Impaired reabsorption of sodium, chloride, and magnesium leads to secondary hyperaldosteronism, hypokalemic metabolic alkalosis, hypomagnesemia, and hypocalciuria. GS manifests with a wide clinical spectrum, ranging from mild or absent symptoms to severe presentations such as muscle weakness or tonic–clonic seizures. Its estimated prevalence is approximately 1 in 40,000 individuals, with higher reported frequencies in Asian populations. 3 Importantly, the hypomagnesemia characteristic of GS can suppress PTH secretion and cause peripheral PTH resistance—mechanisms that typically protect against hypercalcemia.4,5 However, in patients with concurrent PHPT, elevated PTH levels override this protective effect. When coupled with the intrinsic hypocalciuria of GS, this results in marked dysregulation of calcium homeostasis and may precipitate a refractory hypercalcemic crisis characterized by severe hypercalcemia, multiorgan dysfunction, and profound electrolyte abnormalities.
Only a small number of cases describing GS complicated by PHPT have been reported.6–10 Most studies have described patients with GS-related symptoms accompanied by relatively mild hypercalcemia. The present case underscores the severe clinical potential of this coexistence and highlights the diagnostic complexity and therapeutic challenges of managing these overlapping disorders. It represents an exceptionally rare scenario in which a sudden, life-threatening hypercalcemic crisis was the initial presentation, wherein underlying GS contributed to poor responsiveness to standard calcium-lowering therapies.
The reporting of this case adheres to the Case Report (CARE) guidelines. 11
Case presentation
A 69-year-old woman presented with a 3-month history of dizziness, fatigue, and intermittent confusion. Her symptoms began with persistent, unexplained dizziness, profound fatigue, and nausea without vomiting, which persisted despite initial treatment. One week later, she suddenly lost consciousness and required emergency care at a local hospital. Critical laboratory findings at that time (reference ranges provided in Table 1) revealed life-threatening hypercalcemia: serum calcium, 4.84 mmol/L; ionized calcium, 1.63 mmol/L; peak serum creatinine, 106.4 μmol/L; and PTH, 1128 ng/L (notably elevated). She was treated with aggressive fluid resuscitation, diuretics, and urgent hemofiltration. After serum calcium decreased to 2.64 mmol/L, she regained consciousness and appetite; however, hypercalcemia subsequently rebounded. Intravenous administration of 4 mg zoledronic acid reduced her calcium nadir to 2.61 mmol/L. Ultrasound imaging revealed a well-defined hypoechoic nodule (2.19 × 1.21 cm) in the posterior right lobe, suspicious for a parathyroid lesion. Multiple additional hypoechoic nodules (TI-RADS 3–4a) were also identified. Given her critical condition, surgical intervention was deemed unsuitable; instead, calcium-lowering therapy and close monitoring were initiated. Twenty days prior to the current presentation, the patient experienced recurrent severe symptoms, including profound apathy, incapacitating weakness that prevented standing or walking, nausea, abdominal distension, and constipation. Subsequent testing revealed markedly elevated serum calcium (>3.5 mmol/L) and PTH levels (257.4 ng/L). Initial aggressive treatment with fluid replacement, diuresis, and calcitonin proved ineffective, prompting a second intravenous 4-mg dose of zoledronic acid. Serum calcium levels then fluctuated between 2.74 and 2.98 mmol/L. At presentation to our emergency department, laboratory investigations showed the following: serum creatinine, 26 μmol/L; albumin, 39 g/L; alkaline phosphatase, 46 U/L; calcium, 3.40 mmol/L; potassium, 2.7 mmol/L; sodium, 129 mmol/L; and chloride, 93 mmol/L. Treatment included fluid resuscitation, potassium supplementation, and subcutaneous calcitonin (100 IU twice daily). Persistently elevated follow-up calcium level (3.30 mmol/L) necessitated urgent hospitalization. The patient denied the use of thiazide diuretics or calcium/vitamin D supplements and reported no family history of parathyroid disease or GS. Three months ago, she had been diagnosed with deep vein thrombosis (DVT) in the right lower extremity, accompanied by pronounced swelling, which led to inferior vena cava filter implantation. She has been maintained on long-term anticoagulation with oral rivaroxaban (20 mg daily).
Serial laboratory parameters and clinical interpretation.

PTH: parathyroid hormone; TRP: tubular reabsorption of phosphate; TmP/GFR: tubular maximum reabsorption of phosphate per glomerular filtration rate; PHPT: primary hyperparathyroidism; GS: Gitelman syndrome.
The patient was admitted to the ward. On examination, she was alert but apathetic, demonstrating appropriate engagement despite slurred speech and signs of dehydration. Vital signs were stable, with a body mass index of 22.03 kg/m2, blood pressure of 124/68 mmHg, and a pulse rate of 78 bpm. Neurological examination revealed no focal deficits. Muscle strength was graded 4−/5 in both lower limbs. Lower limb examination revealed no edema on the left, with edema present distal to the knee on the right.
A comprehensive laboratory evaluation was performed at admission, with key parameters and their clinical significance summarized in Table 1. Notable findings were as follows: serum calcium, 3.03 mmol/L; phosphorus, 0.47 mmol/L; sodium, 132 mmol/L; potassium, 2.7 mmol/L; magnesium, 0.27 mmol/L; PTH, 25.7 pmol/L; supine direct renin concentration (DRC), 454.6 μIU/mL; aldosterone, 52.3 ng/dL; and pH, 7.517. Urinary measurements were as follows: potassium, 174.14 mmol/24 h; calcium, 8.84 mmol/24 h; phosphorus, 7.58 mmol/24 h; calcium excretion rate, 1.5%; tubular reabsorption of phosphate (TRP), 76.1%; and tubular maximum reabsorption of phosphate per glomerular filtration rate (TmP/GFR), 0.4 mmol/L. Routine blood tests—including liver/kidney function tests, lipid profile, glycated hemoglobin, tumor marker panel, complete immunological profile, serum immunofixation electrophoresis, catecholamine metabolite testing, and pituitary function tests—were within normal limits. Bone metabolic markers showed a slightly elevated β-CrossLaps (CTx) level of 0.69 ng/mL and a low 25-hydroxyvitamin D level of 18.37 nmol/L. Urinary system color Doppler ultrasound revealed bilateral renal sinus nodular dense shadows, suggestive of stones or calcifications. A Technetium-99m MIBI single-photon emission computed tomography (SPECT)/ computed tomography (CT) scan demonstrated a prominent focus of increased radiotracer uptake (approximately 2.4 ×1.1 × 0.9 cm) posteroinferior to the left thyroid lobe, consistent with hyperfunctioning parathyroid tissue. Additionally, a focal area of mild tracer uptake (approximately 2.1 × 1.3 × 1.1 cm) was observed posteroinferior to the right thyroid lobe, raising concern for a hyperfunctioning parathyroid lesion (Figure 1).
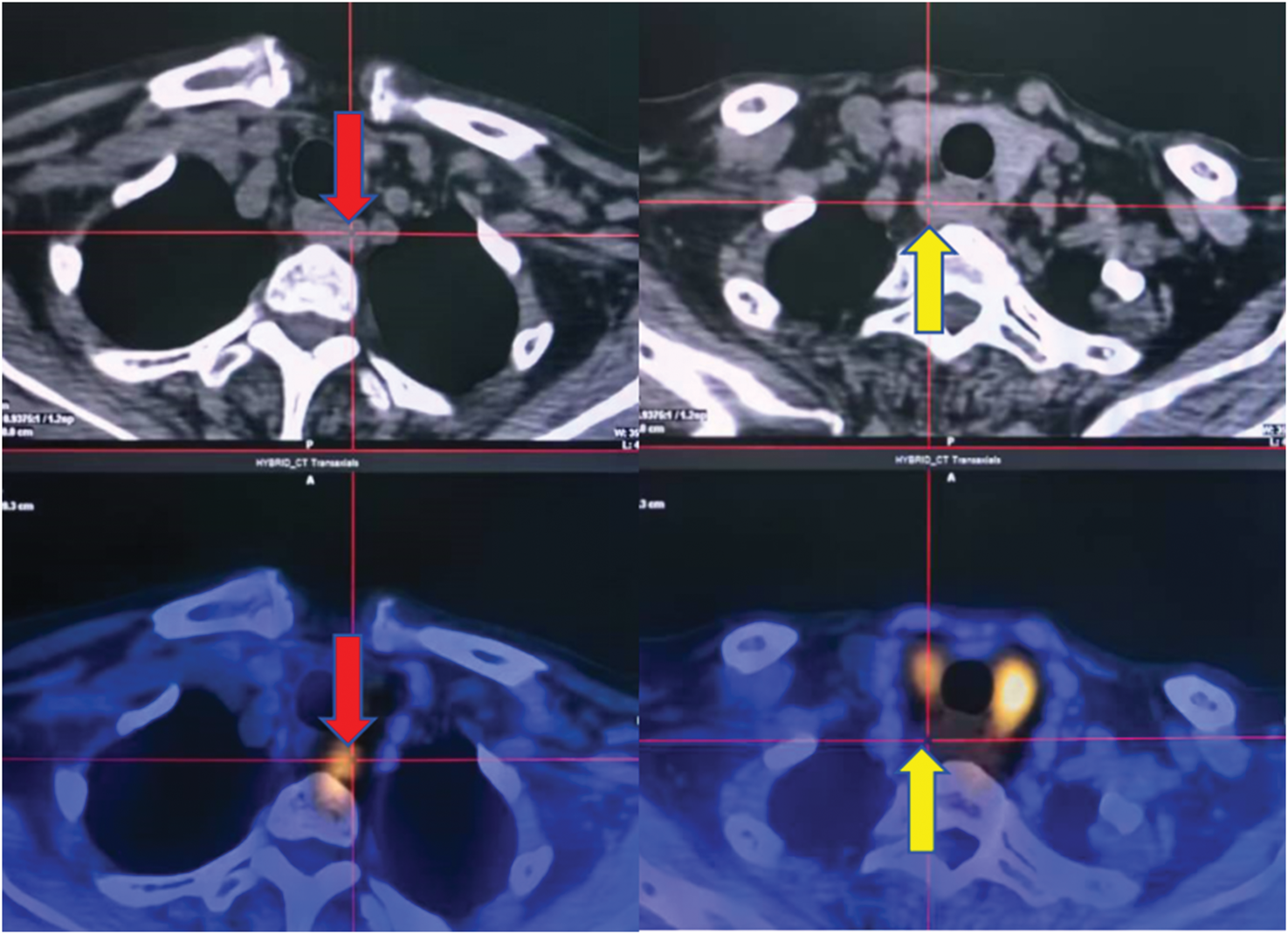
Preoperative Technetium-99m MIBI SPECT/CT imaging of the parathyroid glands. The axial fusion image shows a prominent focus of intense radiotracer uptake (red arrows) posterior to the left inferior thyroid lobe. A second area of mild radiotracer uptake (yellow arrows) is visible posterior to the right thyroid lobe. SPECT/CT: single-photon emission computed tomography/computed tomography.
The patient’s response to calcium-lowering therapy is summarized in Table 2. Initial post-admission management included fluid resuscitation, diuretics, electrolyte supplementation, calcitonin, and anticoagulation. For persistent hypercalcemia unresponsive to this regimen, two sequential doses of denosumab (60 mg each, total 120 mg) were administered, successfully reducing serum calcium to 2.84 mmol/L. Concurrent laboratory values were as follows: serum phosphorus, 0.68 mmol/L; sodium, 137 mmol/L; potassium, 3.4 mmol/L; magnesium, 0.41 mmol/L; and low urinary calcium, 1.36 mmol/24 h. Subsequent bilateral parathyroid exploration revealed encapsulated reddish-brown masses: a 3.0 × 1.5-cm mass at the dorsal lower pole of the left lobe and a 1.5 × 1.2-cm mass at the dorsal midpole of the right lobe. Both lesions were successfully excised. Postoperative pathology confirmed PHPT, showing parathyroid adenomatous hyperplasia. Immunohistochemistry was positive for CK19, PTH, galectin-3, and CgA and negative for calcitonin, synaptophysin, S-100, and TTF-1, with a Ki-67 index of approximately 1%. The patient had pre-existing refractory hypokalemia and hypomagnesemia. A key diagnostic clue was the presence of hypocalciuria prior to normalization of serum calcium, which raised strong clinical suspicion for GS. This suspicion was confirmed by peripheral blood genetic testing, which revealed two heterozygous mutations in the SLC12A3 gene: c.1077C>G (p.Asn359Lys) in exon 8 and c.788_805dup TTGGCGTGGTCTCGGTCA (p.V268_T269insIGVGVSV) in exon 6. Concurrent DNA analysis of the MEN1 and HRPT2 genes ruled out an associated risk of multiple endocrine neoplasia (MEN).
Efficacy of calcium-lowering treatments.

Postoperative care included calcium and calcitriol therapy, initially without potassium or magnesium supplementation. At 3-day follow-up, the following results were recorded: serum calcium, 2.32 mmol/L; phosphorus, 1.41 mmol/L; sodium, 141 mmol/L; potassium, critically low at 2.1 mmol/L; magnesium, severely low at 0.29 mmol/L; and PTH, undetectable. After initiating targeted electrolyte replacement, serum potassium level increased to 3.2 mmol/L and magnesium level to 0.48 mmol/L.
Discussion
PHPT results from excessive secretion of PTH, leading to hypercalcemia, hypophosphatemia, and potential bone or renal complications. Approximately 80%–85% of cases are caused by a solitary adenoma, 10%–15% by hyperplasia, and <1% by carcinoma. In this patient, recurrent, life-threatening hypercalcemic crises—with serum calcium level reaching 4.84 mmol/L and PTH level markedly elevated at 1128 ng/L—were consistent with PHPT. Imaging revealed multiple foci of radiotracer uptake in the parathyroid region, suggesting multigland involvement. Surgical exploration and histopathology confirmed parathyroid adenomatous hyperplasia. Immunohistochemistry was positive for CK19, PTH, galectin-3, and CgA, confirming functional parathyroid tissue, while a low Ki-67 index (∼1%) supported its benign nature.
This case is notable for the co-occurrence of PHPT with GS. Typically, hypomagnesemia in GS confers a protective effect against hypercalcemia by inducing resistance to the calciotropic effects of PTH and active vitamin D as well as impairing renal 1α-hydroxylase activity. Consequently, calcium influx from bone and gut into the circulation is reduced, resulting in hypocalciuria. Therefore, the presence of hypercalcemia in a patient with known or suspected GS should raise suspicion for underlying PHPT. This association provides an important clinical clue in diagnostically challenging cases.
The coexistence of PHPT and GS creates a complex clinical scenario. Despite hypomagnesemia, PTH oversecretion can overcome magnesium-mediated suppression, amplifying end-organ effects. GS-related tubular dysfunction further perturbs calcium homeostasis: hypomagnesemia exacerbates calcium dysregulation, while hypocalciuria—which was present prior to normalization of serum calcium in this case—limits renal calcium excretion, contributing to severe hypercalcemia. The combination of PTH excess and reduced renal calcium excretion can precipitate recurrent, refractory hypercalcemic crises, as observed in this patient. Conventional therapies, including hydration, diuretics, calcitonin, bisphosphonates, or denosumab, often produce suboptimal responses. Severe hypercalcemia can also induce polyuria and electrolyte losses, aggravating dehydration and hypokalemia/hypomagnesemia and triggering secondary hyperaldosteronism. Hypomagnesemia occurs in approximately 25.1% of PHPT patients, 13 sometimes accompanied by mild hypermagnesuria due to reversible tubular defects, 14 increasing the risk of a missed GS diagnosis.
Coexisting PHPT and GS is rare. Unlike previous reports emphasizing GS-related symptoms such as fatigue or muscle cramps in the context of mild hypercalcemia, our case was characterized by recurrent, severe hypercalcemic crises. The classic electrolyte disturbances of GS—hypokalemia, hypomagnesemia, and hypocalciuria—were masked preoperatively by hypercalcemia but were subsequently confirmed genetically. This case highlights that concomitant PHPT should be suspected in GS patients who develop hypercalcemia, particularly when persistent hypokalemia, hypomagnesemia, or hypocalciuria is present. Genetic testing for SLC12A3 is essential for definitive diagnosis.
Management of coexisting PHPT and GS is challenging. Correcting hypomagnesemia may exacerbate hypercalcemia; therefore, PHPT should be addressed first, ideally through parathyroidectomy. Calcium-lowering agents with minimal renal impact, such as denosumab, are preferred. Thiazide diuretics should be avoided due to the risk of worsening hypercalcemia, whereas loop diuretics may be used cautiously in acute settings. Following surgery, PHPT was resolved; however, severe hypokalemia (2.1 mmol/L) and hypomagnesemia (0.29 mmol/L) required aggressive supplementation, confirming GS’s contribution to electrolyte dysregulation. Lifelong monitoring and supplementation are necessary.
The patient’s right lower-limb DVT may have been linked to hypercalcemia, 15 which promotes hypercoagulability through platelet activation, enhanced clotting factor activity, impaired fibrinolysis, 16 and endothelial injury from vascular calcification.17,18 Polyuria associated with hypercalcemia may further concentrate blood, increasing thrombosis risk. Patients with DVT and concurrent hypercalcemia should receive thrombosis prophylaxis while addressing the underlying calcium disorder.
This case underscores the importance of considering rare tubular disorders such as GS in patients with refractory hypercalcemia, especially when accompanied by hypokalemia, hypomagnesemia, or hypocalciuria. Conversely, the development of hypercalcemia in a patient with GS should prompt evaluation for underlying PHPT. Genetic testing can prevent diagnostic delays. Early recognition of combined PHPT and GS is essential. The optimal approach involves surgical management of PHPT, followed by careful correction of GS-related electrolyte abnormalities, which is critical for improving outcomes in these complex cases.
Clinicians should also be aware that PHPT can rarely present with a phenotype mimicking distal renal tubular acidosis, due to hypercalcemia-mediated impairment of urinary acidification. This consideration adds another layer of complexity when evaluating acid–base and electrolyte disturbances in hypercalcemic patients.
Footnotes
Acknowledgments
We are grateful to the patient and her family, who kindly consented to participate in the study. The work was supported by Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-3-002C) and Tianjin Medical University Clinical Special Disease Research Center-Neuroendocrine Tumor Clinical Special Disease Research Center.
Author contributions
MHY, QH, and ML conceptualized and designed the study. MHY collected the data, drafted the initial manuscript, and reviewed and revised the manuscript. BPW, YXL, CGZ, and TL coordinated and supervised data collection and reviewed the manuscript. All authors have read and approved the final version of the manuscript.
Availability of data and materials
The detailed clinical and imaging data are included in the manuscript.
Consent for publication
Informed written consent for the publication of the clinical details and clinical images was obtained from the patient.
Declaration of conflicting interests
The authors declare that they have no conflicts of interest to disclose.
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the General Hospital of Tianjin Medical University. All procedures were conducted in accordance with the tenets of the Declaration of Helsinki. Written informed consent for publication was obtained from the patient.
Funding
Not applicable.