
Abstract
Homocystinuria, an autosomal recessive metabolic disorder, is caused by cystathionine β-synthase deficiency and presents with diverse clinical manifestations, including ectopia lentis, osteoporosis, scoliosis, premature arteriosclerosis, thromboembolism, and intellectual disability. We present a case in which a pedigree was diagnosed with homocystinuria through comprehensive physical examinations, ophthalmologic evaluations, and genetic testing. Three patients carrying a CBS homozygous mutation with an identical genotype presented with varying degrees of ocular and systemic abnormalities. In this pedigree, homocystinuria remained undiagnosed until the affected individuals sought ophthalmologic care for visual impairment. We identified a novel homozygous missense mutation, c.344G>A, in the CBS gene. This pedigree provides valuable insights into the natural progression and prognosis of homocystinuria in the absence of intervention. Genetic testing plays a critical role in elucidating the etiology of this disorder and differentiating it from other conditions. Thus, it should be considered an essential tool for diagnosis and genetic counseling for patients and their family members. Early detection of metabolic disorders can significantly reduce the risk of severe systemic complications by enabling timely and appropriate therapeutic interventions.
Introduction
Homocystinuria, an autosomal recessive metabolic disorder caused by cystathionine β-synthase (CBS) deficiency, is characterized by markedly elevated plasma homocysteine and methionine levels, increased urinary excretion of homocysteine, and reduced concentrations of cystathionine and cysteine in body fluids.1,2 The global prevalence of homocystinuria is estimated at 1 in 100,000 to 1 in 263,000 individuals, based on data from newborn screening and clinically diagnosed cases.3–5 This condition presents with diverse clinical manifestations involving the eyes, skeleton, vascular system, and central nervous system. Key clinical features typically include ectopia lentis, osteoporosis, scoliosis, premature arteriosclerosis, thromboembolism, and intellectual disability.6,7
Previous studies have demonstrated that CBS deficiency disrupts sulfur amino acid metabolism, resulting in ocular complications such as ectopia lentis, high myopia, secondary acute angle-closure glaucoma, optic atrophy, and retinal detachment. 8 Herein, we present a case of a pedigree diagnosed with homocystinuria based on comprehensive physical examinations, ophthalmologic evaluations, and genetic testing. Notably, a novel homozygous mutation, c.344G>A, was identified in the CBS gene. To the best of our knowledge, this is the first report of three homozygous patients within a single pedigree sharing an identical genotype. Observations across different ages provide valuable insights into the natural progression and prognosis of the disorder, underscoring the critical importance of early diagnosis and intervention.
Case presentation
A primary school-aged female child was admitted to the Department of Ophthalmology at the First Affiliated Hospital of Kunming Medical University in August 2023 with a progressive decline in visual acuity and right-eye ophthalmodynia. She was born to nonconsanguineous parents who had no history of ocular abnormalities. There were no complications during pregnancy, delivery, or the neonatal period. Upon examination, her visual acuity was OD: 0.05; OS: 0.05. Refraction showed OD: −20.00 DS (spherical lens) − 2.00 DC (cylindrical lens) × 155° → 0.25; OS: −22.00 DS → 0.3. The intraocular pressure measured via a noncontact tonometer was 24.5 mmHg OD; 12 mmHg OS. The axial lengths were 21.75 mm OD; 22.09 mm OS. A slit-lamp examination revealed conjunctival congestion and corneal edema in the right eye. Additionally, the lens was dislocated, and the pupil was irregularly dilated in the anterior segment. In the left eye, the sclera, conjunctiva, and cornea appeared normal; however, the lens was displaced toward the nasal side, with ruptured zonules curled at the equatorial region (Figure 1). Direct ophthalmoscopy of the posterior segments revealed no significant abnormalities.

Anterior segment photograph of the female proband. The right eye showed dislocation of the transparent lens with an irregularly dilated pupil, while the left eye demonstrated nasal displacement of the lens and curled ruptured zonules at the equatorial region.
The patient was shorter and heavier than the average Chinese girl of the same age, with a height of 130.1 cm (average: 134.1 cm) and a weight of 32.4 kg (average: 28.2 kg). After obtaining informed consent, a comprehensive physical examination was performed, revealing slightly yellowish hair, short and thick fingers, malformed knee joints, and scoliosis of the lumbar spine (Figure 2). Neurological examinations revealed no abnormalities or pathological signs. Laboratory investigations showed a total plasma homocysteine concentration of 3.2 µmol/L (normal: <15 µmol/L) and a methionine concentration of 36.93 µmol/L (normal: 13.08–50.45 µmol/L). Echocardiography did not reveal any anomalies. The constellation of findings, including ectopia lentis, short stature, short and thick fingers, joint abnormalities, and high myopia, along with unremarkable laboratory investigations and echocardiography, was suggestive of Weill–Marchesani syndrome.

Physical examination of the female proband revealed slightly yellowish hair (a), short and thick fingers (b), and malformed knee joints (c).
A comprehensive medical history of the patient’s siblings revealed ocular and systemic abnormalities. Her older brother, who was in his early 20s, underwent surgeries for suspensory intraocular lens (IOL) implantation in both eyes at the age of 7 years because of complete lens dislocation. He exhibited slightly yellowish hair and mild intellectual delay, without significant bone or joint abnormalities (Figure 3). His height was 171 cm (average: 169.7 cm) and weight 68.1 kg (average: 66.2 kg), consistent with a normal body build. Laboratory investigations revealed a total plasma homocysteine concentration of 15.3 µmol/L and a methionine concentration of 54.62 µmol/L. Her preschool-aged younger brother presented with myopia and partial lens dislocation on cycloplegic refraction, and slack zonules and subluxated lenses were observed upon examination (Figure 4). Refraction measurements showed OD: −4.00 DS −2.50 DC × 65° → 0.25; OS: −8.50 DS −2.50 DC × 90° → 0.12. Physical examination revealed slightly yellowish hair, short and thick fingers, and malformed knee joints (Figure 5). The laboratory results revealed a markedly elevated total plasma homocysteine concentration of 33.6 µmol/L and a methionine concentration of 98.66 µmol/L. Echocardiography assessments revealed no anomalies in the siblings. Additionally, no other family members were diagnosed with similar ocular abnormalities during childhood.

Portrait of the proband’s older brother (a) showing his thick fingers (b).

Anterior segment photograph of the proband’s younger brother. Partial lens dislocation and slack zonules were observed in both eyes after dilation.
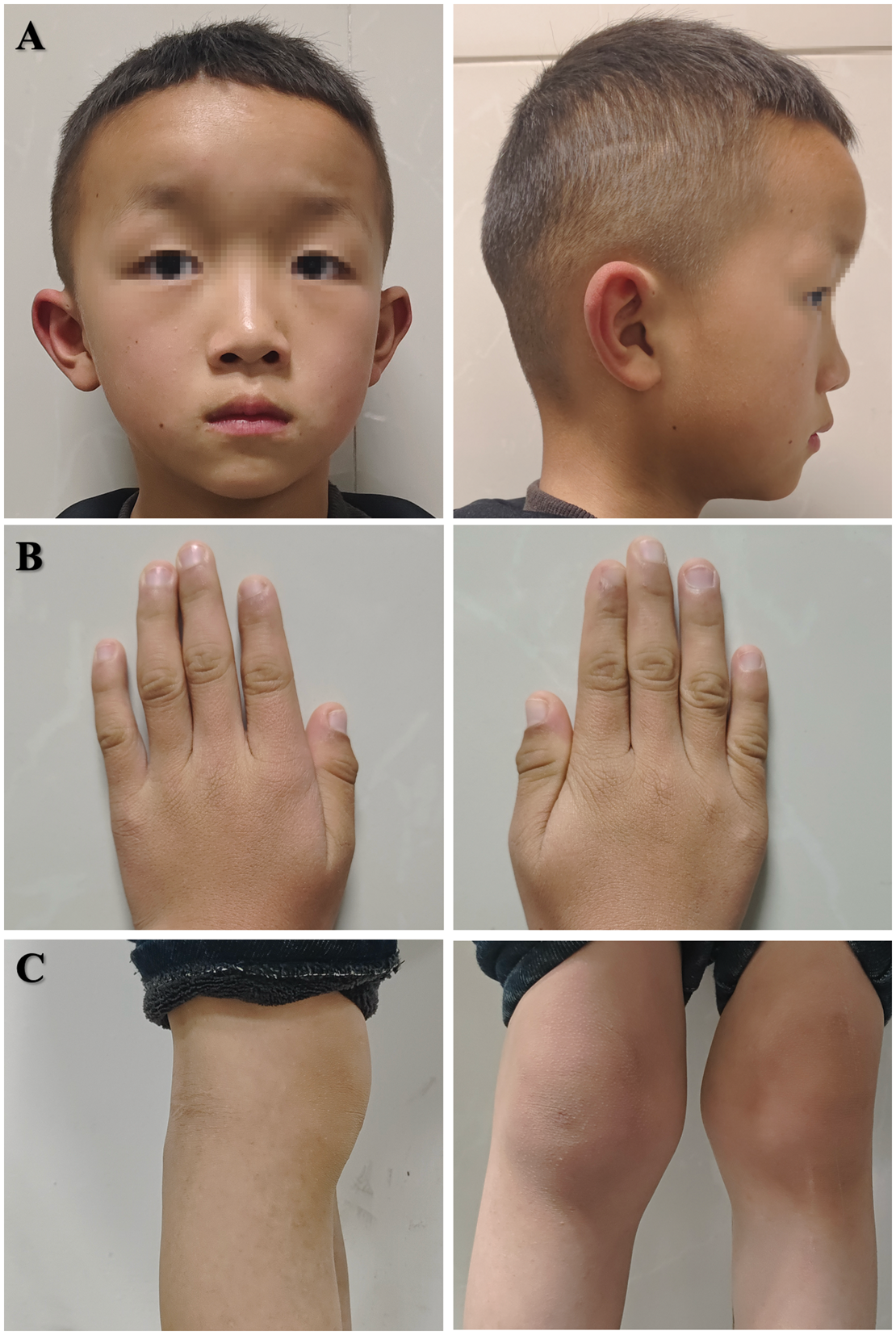
Physical examination of the proband’s younger brother revealed slightly yellowish hair (a), short and thick fingers (b), and malformed knee joints (c).
The patient and her family members underwent whole-exome sequencing (WES), which revealed a novel homozygous missense mutation in the CBS gene. This mutation was subsequently confirmed by Sanger sequencing (Figure 6). Its pathogenicity was predicted using Combined Annotation-Dependent Depletion (CADD, https://cadd.gs.washington.edu/, version 1.4), Rare Exome Variant Ensemble Learner (REVEL, https://sites.google.com/site/revelgenomics/), Polymorphism Phenotyping v2 (PolyPhen2, http://genetics.bwh.harvard.edu/pph2/), Sorting Tolerant From Intolerant (SIFT, http://sift.jcvi.org), and Protein Variation Effect Analyzer (PROVEAN, http://provean.jcvi.org/genome_submit_2.php). The allele frequency of this mutation was not found in the Genome Aggregation Database (https://gnomad.broadinstitute.org) (Table 1). Therefore, the diagnosis of homocystinuria was established by correlating the genetic findings with the clinical manifestations observed in the proband and her family members.

Sanger sequencing confirmed the CBS gene homozygous missense mutation identified in this family, which is also depicted in the pedigree.
CBS mutation identified in this study (Ref. NM_000071.2).

CADD: Combined Annotation-Dependent Depletion; REVEL: Rare Exome Variant Ensemble Learner; Polyphen2: Polymorphism Phenotyping v2; SIFT: Sorting Tolerant From Intolerant; PROVEAN: Protein Variation Effect Analyzer; gnomAD AF: Genome Aggregation Database Allele Frequency; PRD: probably damaging; DA: damaging; DE: deleterious; /: not applicable.
The patient underwent lens extraction combined with anterior vitrectomy, followed by suspension of a posterior chamber IOL through scleral incisions in the right eye (Figure 7). A similar procedure was performed in the left eye 1 month later. Six months postoperatively, her best-corrected visual acuity improved to OD: 0.6; OS: 0.5. The slit-lamp examination revealed a transparent cornea, clear anterior chamber, and well-positioned IOL (Figure 8).

Diagram and surgical process of posterior chamber intraocular lens suspension through scleral incisions in the right eye.
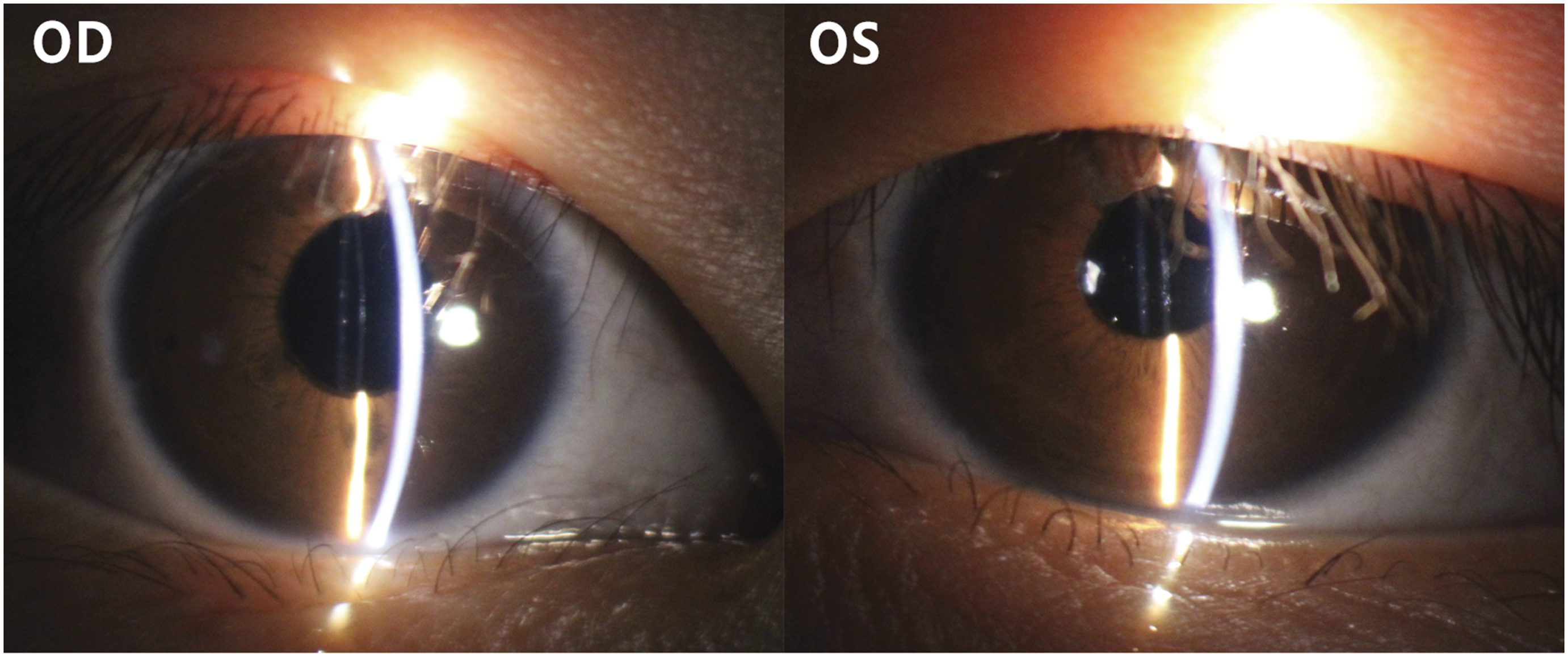
Anterior segment photograph of the female proband 6 months after surgery, showing a transparent cornea, clear anterior chamber, and appropriately positioned intraocular lens.
All procedures in this study conformed to the tenets of the Declaration of Helsinki. Informed consent was obtained from all participants or their guardians prior to the collection of clinical data and venous blood samples. All patient details were de-identified. This case report was written in accordance with the Case Report (CARE) guidelines. 9
Discussion
In this study, we diagnosed a pedigree with homocystinuria, in which the proband presented with ectopia lentis accompanied with ocular symptoms of high myopia. The patient’s characteristics, including short stature, brachydactyly, malformed knee joints, scoliosis of the lumbar spine, and normal concentrations of homocysteine and methionine, initially suggested a differential diagnosis of Weill–Marchesani syndrome. WES followed by Sanger sequencing revealed a novel homozygous missense mutation (c.344CG>A) in the CBS gene, despite the absence of consanguinity in her family.
In this pedigree, homocystinuria remained undiagnosed until the affected individuals presented to ophthalmologists with visual impairment. Currently, homocystinuria is not routinely included in neonatal screening programs in most regions. Therefore, genetic testing plays a critical role in providing genetic counseling, elucidating the etiology of the disorder, and differentiating it from conditions such as Weill–Marchesani syndrome and Marfan syndrome, particularly when isolated ectopia lentis is observed as a shared feature. These findings provide insights into the natural progression and prognosis of the disorder in the absence of intervention. Additionally, genetic diagnosis provided valuable guidance for ophthalmic interventions and nutritional therapy.
Mutations in the CBS gene can result in ocular complications secondary to homocystinuria, including ectopia lentis, high myopia, secondary acute angle-closure glaucoma, optic atrophy, and retinal detachment. Patients with homocystinuria typically present with Marfanoid habitus, characterized by arachnodactyly. In this case, none of the affected individuals exhibited Marfanoid features, despite variations in plasma homocysteine and methionine concentrations. The phenotypic heterogeneity observed among patients with the same genotype may indicate the involvement of other regulatory factors or differences in dietary methionine intake. Further investigations are warranted to clarify the underlying mechanisms. Notably, the total plasma homocysteine and methionine concentrations of the proband were within normal ranges and did not vary with age in this pedigree, despite denying nutritional therapies or any specific dietary preferences. Cardiovascular abnormalities were absent in these patients, and bone mineral density assessments lacked diagnostic significance in underage siblings. Consequently, genetic testing established the definitive diagnosis and elucidated the underlying etiology in this case, thereby highlighting its importance as a fundamental tool for diagnosing pediatric patients with ocular diseases.
Previous studies have predominantly identified missense mutations in patients with homocystinuria.3–5,10 Early and long-term pyridoxine therapy has been shown to significantly reduce homocysteine concentrations in patients with specific mutations.11–13 This reduction helps prevent the progression of myopia and ectopia lentis and mitigate cardiovascular risks and skeletal abnormalities. If left untreated, ectopia lentis tends to progressively worsen, with ocular complications becoming more severe with aging, as observed in the three patients in this study. Partial lens dislocation was detected during cycloplegic refraction in the youngest patient, whereas complete lens dislocation was observed on slit-lamp examination in the older siblings.
In conclusion, these findings underscore the critical importance of early diagnosis and timely clinical intervention in homocystinuria. Ophthalmic surgical interventions and nutritional therapies are essential for improving visual acuity and preventing life-threatening systemic complications in these patients.
Footnotes
Acknowledgments
The authors sincerely thank all participants and their family members for their involvement in this study and for granting permission to use their clinical data.
Author contributions
Xingyu Xu drafted the manuscript and analyzed the data. Zihan He and Yiwei Shen contributed to data collection. Jianfeng Zhao and Yu Geng revised the manuscript for intellectual content. Yu Geng performed the surgeries. All authors approved the final manuscript.
Consent for publication
Written informed consent was obtained from the participants or their guardians for publication of their clinical details along with images in this study.
Data availability statement
All data in this study are available from the corresponding author upon reasonable request.
Declaration of conflicting interest
The authors declare that they have no competing interests.
Ethics approval
All procedures in this study conformed to the tenets of the Helsinki Declaration. Informed consent was obtained from all participants or their guardians prior to the collection of clinical data and venous blood samples. Formal ethics committee approval was not required for this case report.
Funding
This study was supported by grants from the Yunnan Province Science and Technology Department (202301AU070166 and 202405AC350057) and the First Affiliated Hospital of Kunming Medical University.