
Abstract
Smith-Magenis syndrome (SMS) and Dandy-Walker malformation (DWM) are uncommon genetic conditions with nonspecific clinical features, which makes reaching a definitive diagnosis challenging. We describe here, a 2-year-old girl who was diagnosed with SMS at the age of 12 months due to delayed growth and development. The child presented to hospital with acute heart failure and respiratory failure. During the treatment process, her response was limited, and her recovery was slow. A subsequent head computed tomography (CT) scan showed abnormalities consistent with the diagnosis of comorbid DWM. We believe that this is the first reported case of a patient with SMS combined with DWM. By reporting this case, we aim to offer clinicians valuable insights into these rare diseases and provide a framework for future clinical diagnosis and treatment.
Background
Smith-Magenis syndrome (SMS) is a clinical syndrome of multiple congenital abnormalities due to microdeletions or heterozygous mutations on chromosome 17p11.2, which contains the retinoic acid-induced 1(RAI1) gene. 1 SMS was first identified in the USA in the 1980s, and occurs in approximately 1 in 15,000 to 25,000 births.1,2 SMS is mainly characterized by delayed intellectual and motor development, growth retardation, sleep disorders, behavioural abnormalities, special facial features, and skeletal malformations. 1 Dandy-Walker malformation (DWM) or syndrome is a rare congenital central nervous system (CNS) disorder that affects development of the cerebellum and has a prevalence of approximately 1 in 350,000 live births in the USA. 3 Most patients present with signs and symptoms of increased intracranial pressure. 3 Patients may also have malformations of the heart, face, limbs, and gastrointestinal or genitourinary system. 3
The coexistence of SMS with DWM is extremely rare. We present here, the case of a 2-year-old girl previously diagnosed with SMS a year earlier, who was admitted to the hospital with congenital heart malformation and acute heart failure triggered by a pulmonary infection. During the course of her subsequent diagnosis and treatment she was found to have concomitant comorbidity with DWM.
Case report
A 2-year-old girl, with a birth weight of 3.2 kg, born via spontaneous delivery at term, was admitted to the paediatric intensive care unit (PICU) for chest tightness, shortness of breath, and generalized oedema. A ventricular septal defect was detected at birth, and dynamic observation had been recommended. During her subsequent growth and development, the family had observed that the child's motor and language development was delayed. She was able to walk independently, though with an unsteady gait, and could only produce simple sounds. She had trouble falling asleep, was easily awakened during the night, and exhibited irritability. Furthermore, she exhibited reduced pain sensitivity, a tendency to pick her nose, experienced a decline in hearing, and was often prone to constipation. Her parents denied any history of seizures or similar conditions in the family. Twelve months previously she had a genetic test following several unsuccessful hearing tests. Results suggested that the infant had a 17p11.2 (chr17:16836379_20355202) x1 deletion with a deletion length of 3.52 Mb. (Figure 1(d)). The related genes were RAI1, MYO5A, TOP3A, TNFRSF13B, FLCN, GID4, ALKBH5. Based on clinical manifestations, the child was diagnosed with SMS.
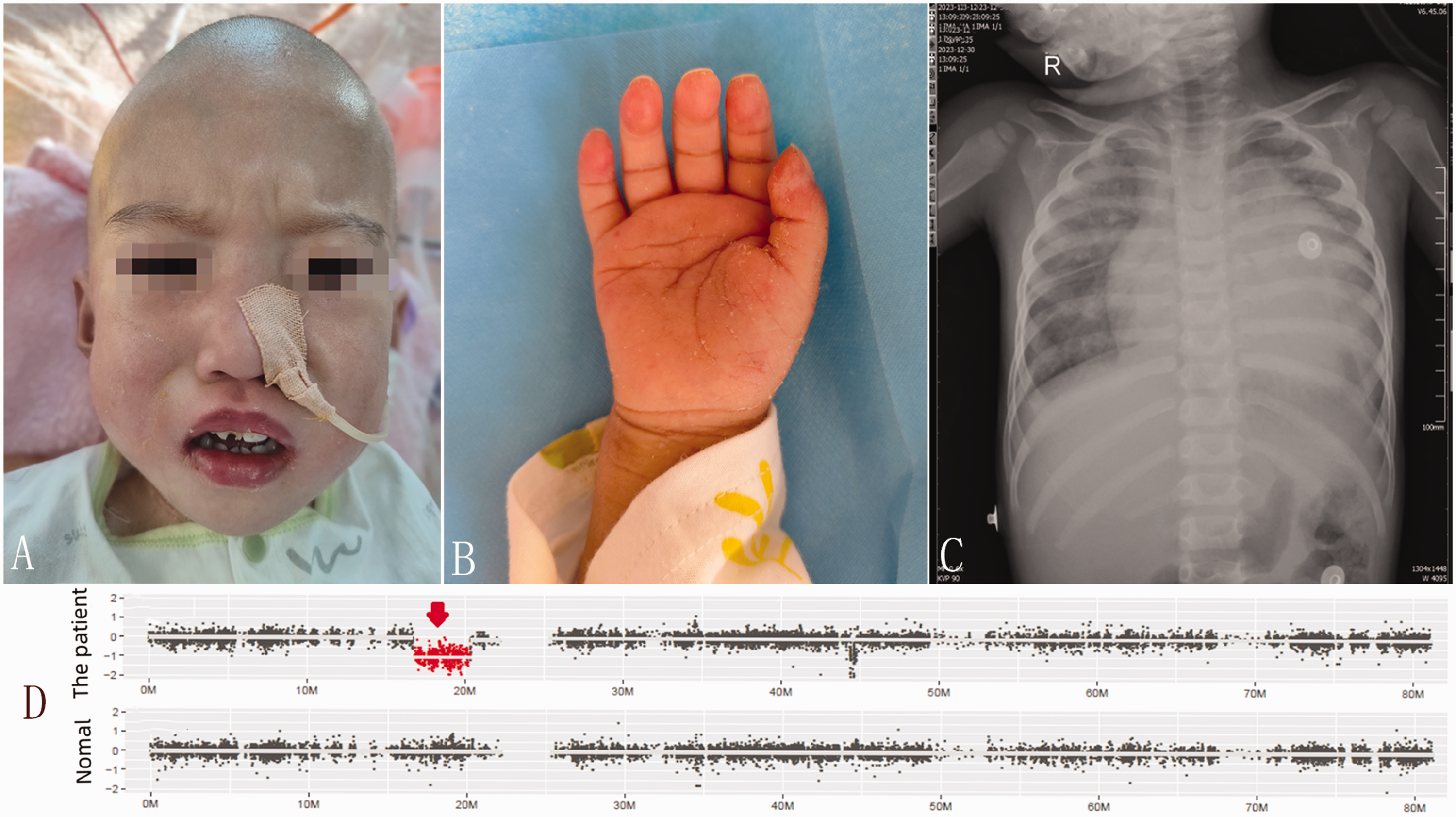
(A) The patient had peculiar facial features (i.e., prominent forehead, flat face, wide eye spacing, low nasal bridge, downward corner of the mouth, mild ectropion of the upper lip, and enamel hypoplasia). (B) Her fingers were short, and her palm was short and wide. (C) X-ray showed enlarged cardiac shadow, thickened and disordered lung markings, scattered patchy high-density shadows in both lungs, primarily in the right lung and (D) results of genetic testing. The red arrow marked area indicates the patient’s gene deletion fragments.
On admission to hospital, she presented with cyanotic lips, puffy eyelids and peculiar facial features (Figure 1(a)). She had a prominent forehead, flat face, wide eye spacing, low nasal bridge, downward corner of the mouth, mild ectropion of the upper lip, and enamel hypoplasia. Coarse breath sounds were detected in both lungs, with audible wet rales. She had enlarged cardiac boundaries, and pathological systolic blowing-like murmurs could be heard between the left sternum and 3-4 ribs. Her fingers were short and her, palms were short and wide (Figure 1(b)). Both lower limbs were oedematous. The muscle strength of all four limbs was grade 4, and bilateral knee jerks were not elicited. Laboratory test results showed elevated white blood cells (WBCs), neutrophils, procalcitonin and B-type natriuretic peptide (NT-proBNP; >35,000 pg/ml). ECG showed sinus tachycardia and frequent ventricular preterm contractions. Chest X-ray showed an enlarged cardiac shadow, thickened and irregular lung markings, along with scattered patchy high-density opacities in both lungs, primarily in the right lung (Figure 1(c)). Ultrasonic cardiography showed: congenital heart disease: ventricular septal defect (VSD); bi-directional shunts at the ventricular level; enlargement of the whole heart; mitral regurgitation (moderate); tricuspid regurgitation (severe); pulmonary regurgitation (mild); left ventricular ejection fraction (LVEF), 31%; pulmonary arterial hypertension (PAH; systolic pressure, >70 mmHg) (Figures 2(a), (b), and (c)). The infant was diagnosed with acute heart failure, congenital heart malformation, ventricular septal defect, severe pneumonia, and respiratory failure.

(A) Ventricular septal defect (VSD) was seen in the short-axis section of the great arteries on ultrasonic cardiography (AO, aorta; PA, pulmonary artery). (B & C) Cardiac colour doppler ultrasound showed mitral and tricuspid regurgitation in the apical four-chamber view (white arrow). (D) Computed tomography (CT) scan of the head (sagittal plane) showing hypoplasia of the cerebellar vermis and cystic dilatation of the fourth ventricle. (E) CT scan of the head (transverse plane) showing enlarged posterior fossa and cyst on the left side of the cisterna magna and (F) CT scan of the head showing dilatation of the cerebral and third ventricles, enlarged posterior fossa, and hydrocephalus.
After receiving antibiotic treatment, cardiac support, and management of pulmonary arterial hypertension (PAH), the child underwent repair of the VSD, along with tricuspid and mitral valvuloplasty, in the cardiac surgery department. Following surgery, her limb muscle strength and mental response were observed to have decreased significantly compared with the preoperative period. Her limb muscle strength was now grade 2, and it was difficult to wean her off the mechanical ventilator. To clarify if intracranial lesions were present, a head computed tomography (CT) scan and an electroencephalogram (EEG) test were performed.
Head CT scans showed a cyst in the posterior cranial fossa linked to the fourth ventricle, a small cerebellar vermis, and supratentorial hydrocephalus (Figures 2(d), (e), (f)). A diagnosis of DWM was considered. The EEG showed predominantly slow wave activity and sleep waves (i.e., sleep staging was unclear). To support the development of an effective treatment plan, the infant underwent a multidisciplinary consultation involving neurology, nutrition, clinical pharmacy, and child health care, and her family were encouraged to actively participate in her rehabilitation therapy. The infant’s muscle strength gradually improved, and she was taken off the ventilator on the 10th day after the operation. She continued rehabilitation therapy for 35 days, after which she was discharged from the hospital. The child continued with regular rehabilitation outside the hospital and showed good recovery at the three-month postoperative follow-up.
The reporting of this study conforms to CARE guidelines. 4 The child's family provided consent for publication of her anonymised data.
Discussion
SMS is a complex genetic disorder, with approximately 90% of patients having a deletion of the 17p11.2 fragment and the remaining 10% having mutations in the RAI1 (retinoic acid-induced 1) gene. 1 Multiple genes are known to be localized in the chromosome 17p11.2 deleted region, of which deletion of the RAI1 gene may be a key gene in the development of SMS. 1 Importantly, the RAI1 gene encodes retinoic acid-inducible protein 1, which is involved in the development of multiple systems. 5 The gene is widely expressed in the central nervous system (CNS) and may be involved in a variety of disorders such as schizophrenia, autism, cerebellar ataxia. 6 Although there is a great deal of overlap, some phenotypic differences have been reported between patients with 17p11.2 deletion and those with mutations in RAI1. 7 Patients with 17p11.2 deletion exhibit a high prevalence of cardiac and renal abnormalities, motor delays, short stature, and hearing impairment. 8 By contrast, patients with RAI1 mutations have an increased risk of overweight/obesity, feeding disorders, poly-embolism, muscle spasms, and dry skin. 9
SMS is characterized by a clinically recognizable phenotype that includes physical, developmental, neurological, and behavioural features.1,10,11 Physical features include distinctive craniofacial anomalies (e.g., microcephaly, thick hair, low hairline, broad forehead). In addition, patients may have skeletal malformations (i.e., short stature, scoliosis, short limbs, short fingers, and toes). Most patients have developmental delays and cognitive disorders (e.g., feeding difficulties in the neonatal period, growth stagnation, motor retardation in children, cognitive dysfunction, mild-to-moderate mental retardation). Behavioural abnormalities include: attention deficit; maladaptive disorders; self-injurious behaviours (i.e., head-banging, self-biting, jacking, nail picking). Patients also tend to have sleep abnormalities (e.g., difficulty in falling asleep, frequent nocturnal awakenings, early awakenings, decreased rapid eye movement (REM) sleep, enuresis, snoring, excessive daytime sleepiness). They may also exhibit stereotypical behaviours, such as intermittent upper-body squeezing or self-hugging. Otolaryngological problems (e.g., hearing loss, auditory hypersensitivity) and ocular abnormalities (e.g., ocular lesions: refractive error, small cornea) are common. Other systems that could show abnormalities include the cardiovascular, renal/urinary, metabolic, immune, and dermatological systems.1,10,11 Significantly, many features seen in SMS can also present in other genetic syndromes, potentially leading to diagnostic challenges.
Diagnosis of SMS should be based upon initial clinical suspicion of the disorder followed by a molecular confirmation of the chromosomal/gene defect. An example of a diagnostic flowchart is shown in Figure 3. Currently, there is no specific treatment for SMS and its management focuses on treating the medical issues presented by the affected individual.1,12 Since almost every patient with SMS has sleep disorders and behavioural abnormalities, treatment has tended to focus on sleep improvement and intervention of abnormal behaviour. 12 Appropriate assessment of relevant problems, early targeted intervention, and rigorous maintenance therapy can improve the overall health status, quality of life, and social functioning of patients. 12 The prognosis depends on the severity of the organ or tissue damage and whether the disease is diagnosed and treated early.

A flowchart for the diagnosis of Smith-Magenis syndrome (SMS). CGH, comparative genomic hybridization.
DWM is an uncommon congenital CNS malformation with unclear aetiological factors, and it is considered a multifactorial disorder. 3 The syndrome is mostly sporadic and isolated, but can also occur as a part of single-gene disorders, genetic syndromes, or chromosomal abnormalities.3,13 Environmental factors, including vertical transmission of viruses, maternal diabetes, and exposure to alcohol, drugs, and toxins during pregnancy, also play a role in the development of the disease. 13 In addition, a few cases of de-novo mutations have also been reported. 13 With the exception of SMS, several genetic syndromes have been reported to be associated with DWM.3,14 They can broadly be grouped into Mendelian disorders (e.g., Joubert–Boltshauser syndrome, Warburg syndrome), chromosomal aberrations (e.g., PHACE syndrome, trisomy 18 syndrome), disorders associated with exposure to environmental agents and sporadic syndromes (e.g., Klippel–Feil syndrome). The clinical features of DWM are non-specific and primarily associated with cerebellar structural abnormalities and intracranial hypertension resulting from hydrocephalus and an enlarged posterior fossa cyst. Patients may present with motor developmental delay, seizures, speech dysfunction, motor incoordination, psychiatric symptoms (e.g., bipolar disorder, depression, schizophrenia), and symptoms of intracranial hypertension (e.g., vomiting, lethargy, irritability and in severe cases, altered levels of consciousness). The syndromic form of DWM may also have malformations of the heart, face, limbs, and gastrointestinal or genitourinary system. 3 The clinical manifestations may exist alone or in combination.
Imaging plays a key role in the diagnosis of DWM. 3 The diagnosis of DWM is primarily based on its four characteristic manifestations which are: complete or partial agenesis of the cerebellar vermis; cystic dilation of the 4th ventricle; enlargement of the posterior fossa; hydrocephalus. When DWM is suspected, chromosomal microarray analysis (CMA) should also be recommended as a diagnostic test. An example of a diagnostic flowchart is shown in Figure 4. The main therapeutic goal is the management of hydrocephalus and posterior cranial fossa cysts usually through surgery. 3 Surgical treatment may include ventriculoperitoneal or cystoperitoneal shunts or posterior cranial fossa cystectomy. While surgery may improve outcomes in some patients, the overall prognosis for individuals with DWM remains poor. 3 The prognosis is dependent on several factors, including the severity of the malformation, the promptness of diagnosis and intervention, the presence of associated complications and the presence of concomitant genetic syndromes.
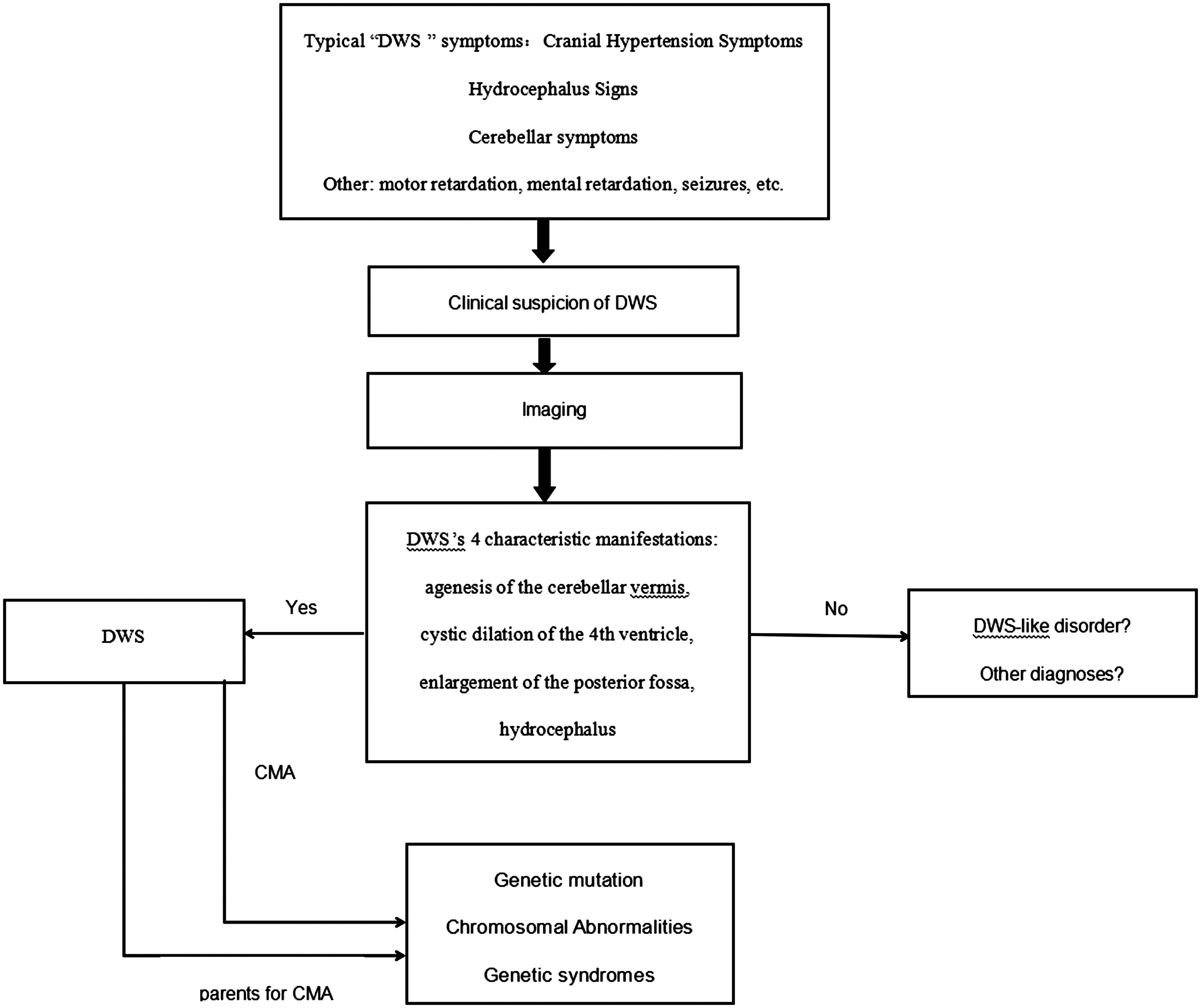
A flowchart for the diagnosis of Dandy-Walker syndrome (DWS) or malformation. CMA, chromosomal microarray analysis.
To the best of our knowledge this is the first report of a patient with SMS and DWM. SMS and DWM are both rare conditions with clinically nonspecific manifestations. Both conditions may present with growth retardation and may be accompanied by other systemic malformations (e.g., neurological, cardiovascular). Therefore, patients may present with a variety of symptoms, which can obscure signs of the disorders and complicate the diagnosis. This article describes the case of a child who was admitted to hospital with acute heart failure induced by a severe pulmonary infection. Through a combination of physical examination and comprehensive diagnostic tests, the patient was ultimately diagnosed with SMS in conjunction with DWM during the course of diagnosis and treatment. Using this patient's treatment as an example, we emphasize that the treatment approach should be tailored based on the severity of the presenting symptoms. For example, our patient was admitted to hospital with heart failure and respiratory failure, so the treatment goal at that time was to improve heart function and correct respiratory failure. With improvement of heart function, the child showed a delayed response, poor strength, and difficulty in weaning off the ventilator. At that time, the focus of the treatment changed to respiratory function and limb function exercise. Following improvement in her respiratory function, and after she had been weaned off the ventilator, the focus of treatment changed to sleep, language, and behavioural interventions. After discharge, the patient required annual comprehensive clinical and diagnostic assessments to achieve effective management. By presenting this case, we aim to help clinicians recognize these two rare diseases, enabling early diagnosis and treatment to improve patient outcomes.
Footnotes
Acknowledgements
We would like to thank Juan Wu and Wanjun Ma for their help in processing the cardiac ultrasound and cranial CT images.
Declaration of conflicting interests
The authors declare that there are no conflicts of interest.
Funding
This research received no specific grant from a funding agency in the public, commercial, or not-for-profit sectors.