
Abstract
Objective
To investigate the effects of hydrogen therapy on epileptic seizures in rats with refractory status epilepticus and the underlying mechanisms.
Methods
Status epilepticus was induced using pilocarpine. The effects of hydrogen treatment on epilepsy severity in model rats were then monitored using Racine scores and electroencephalography (EEG), followed by western blot of plasma membrane N-methyl-D-aspartate receptor subtype 2B (NR2B) and phosphorylated NR2B expression. We also generated a cellular epilepsy model using Mg2+-free medium and used polymerase chain reaction to investigate the neuroprotective effects of hydrogen.
Results
There were no significant differences in Racine scores between the hydrogen and control groups. EEG amplitudes were lower in the hydrogen treatment group than in the control group. In epilepsy model rats, hippocampal cell membrane NR2B expression and phosphorylation increased gradually over time. Although hippocampal cell membrane NR2B expression was not significantly different between the two groups, NR2B phosphorylation levels were significantly lower in the hydrogen group. Hydrogen treatment also increased superoxide dismutase, mitochondrial (SOD2) expression.
Conclusions
Hydrogen treatment reduced EEG amplitudes and NR2B phosphorylation; it also decreased neuronal death by reducing oxidative stress. Hydrogen may thus be a potential treatment for refractory status epilepticus by inhibiting membrane NR2B phosphorylation and oxidative stress.
Keywords
Introduction
Most epileptic seizures last less than 5 minutes; however, when seizures last for more than 5 minutes, it becomes difficult for them to terminate spontaneously, and the disease can progress to status epilepticus (SE).1,2 In 12% to 43% of patients with SE in intensive care units, the disease progresses to refractory SE (RSE), and in 10% to 15% of these patients, the disease further progresses to super-RSE. 3 Seizures that last longer are more harmful to patients. Sustained epileptiform discharges can cause inflammation, oxidative stress, and electrolyte and acid–base imbalances, thus leading to neuronal death and varying degrees of neurological impairment in patients. Approximately two-fifths of RSE patients eventually die of RSE-related complications. Even when patients survive, serious neurological deficits—such as refractory epilepsy or cognitive dysfunction—persist. The current aim of SE treatments is to terminate epileptic seizures as soon as possible via symptomatic and supportive therapies.
Benzodiazepines are widely used as first-line agents for the treatment of SE because of their high safety and ease of use. However, if epileptic seizures continue, gamma-aminobutyric acid-A receptors (GABAARs) in the postsynaptic membranes of hippocampal neurons are internalized, and the numbers of functionally active GABAARs in the synaptic membrane rapidly decrease. This leads to a rapid decline in the efficacy of benzodiazepines and phenobarbital drugs, and the subsequent development of RSE.4–7 By contrast, persistent seizures can cause the numbers of functionally active N-methyl-D-aspartate (NMDA) receptors in the synaptic membrane to gradually increase; thus, drugs that act on NMDA receptors may be effective for treating RSE. 8 Clinically, the NMDA receptor antagonist ketamine has shown efficacy in the treatment of RSE.9,10 The use of this treatment should be based on time-dependent changes in neurotransmission during SE.
Hydrogen can diffuse freely, pass quickly through the blood–brain barrier and cell membranes, enter tissues and nerve cells, and exert unique and selective antioxidant and anti-inflammatory effects; furthermore, it has no side effects.11–14 When the hydrogen concentration in arterial and venous blood reaches approximately 10 to 20 M, it has a good therapeutic effect without any side effects. 14 Moreover, after the supply of hydrogen is stopped, the blood hydrogen concentration drops rapidly. In arterial blood, the hydrogen concentration drops to 10% of the original concentration after approximately 6 minutes; in venous blood it takes slightly longer (approximately 18 minutes). 14 Our earlier research revealed that hydrogen administration enhances the cognitive abilities of rats with epilepsy, and suggested that the underlying process involves a hydrogen-induced decrease in necroptosis in hippocampal neurons. Nonetheless, the potential therapeutic impact of hydrogen on RSE remains uncertain. Several studies have reported that hydrogen can regulate the function of NMDA receptors by regulating membrane transport of the NMDA receptor subtype 2B (NR2B) subunit, thereby reducing nerve excitability.15–17 These results suggest that hydrogen is a potential new treatment for RSE. To date, however, there are no detailed reports of the use of hydrogen to treat RSE.
In the present study, we aimed to investigate the therapeutic effects of hydrogen in RSE and their possible mechanisms. First, we screened an appropriate animal model of RSE. Second, we observed whether hydrogen had therapeutic effects on epileptic seizures in RSE and undertook a preliminarily exploration of their mechanisms. Finally, we investigated the therapeutic effects of hydrogen on RSE-induced neuronal death and the possible underlying mechanisms in a cellular model of epilepsy.
Materials and methods
In vivo experiments
Electrode implantation
All electrodes and fixation nuts were placed in 75% ethanol solution for later use. The rats were anesthetized by intraperitoneal administration of 2% pentobarbital. After anesthesia, the head of each rat was shaved, and the rat was subsequently transferred to a stereotactic system. An ear rod was inserted into the external auditory canal of the rat and fixed; this was followed by the insertion of another ear rod into the other ear canal. Bilateral ear rod symmetry was necessary to ensure that the rat head was maintained in the midline position. The ear rod was then fixed into place, and the height was adjusted so that the skull was in a horizontal position. Next, the skin of the head was disinfected with 75% alcohol. The skin was lifted with tweezers, and the skin and muscle fascia of the head were cut along the midline to fully expose the operation site, which was repeatedly scrubbed with 3% hydrogen peroxide. The position of the electrode was determined according to Paxinos and Watson’s stereotactic atlas. 18 After localization, the left and right positioning points were marked (hippocampal recording electrode site: anterior/posterior: −3.20 mm, lateral/medial: 1.80 mm, dorsal/ventral: 3.0 mm, bilaterally symmetrical). The skull was then carefully opened with a dental drill, and the dura mater was gently torn with a needle. Subsequently, a minor channel was created where the binocular connection line intersected with the midline of the skull, to insert the skull reference electrode. A fixation nut was placed on the bare skull surface external to the electrode implantation site, and the electrode was then placed. Initial fixation was performed with 502 glue, and refixation was performed with dental cement. To prevent brain damage, we ensured that the nuts were not installed too deep. Each rat was then placed on a constant-temperature blanket to maintain warmth, and was returned to the cage for normal feeding after they had fully awakened. After electrode placement, the rats were maintained for 1 week to recover.
During the experiment, the rats were kept in an environment free of pathogens and had access to food and water at all times. They were maintained on a 12-hour light/dark cycle at a temperature of 20°C to 22°C. All animal experiments were performed in accordance with the National Institutes of Health (NIH) guidelines for using experimental animals, and were approved by the Xijing Hospital of Fourth Military Medical University’s Animal Experiment Administration Committee (XJ-20220432). All methods are reported in accordance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) 2.0 guidelines for animal studies.
Electroencephalography (EEG) recordings and behavioral analyses
After electrode implantation and 1 week of recovery, the RSE model was established, and EEGs were recorded. After the experiments, another researcher transformed and analyzed the data from each group using MATLAB (MathWorks, Natick, MA, USA) in accordance with the principles of double blinding. Each rat received an intraperitoneal injection of lithium chloride (180 mg/kg). At 18 to 20 hours after the injection of lithium chloride, an intraperitoneal injection of pilocarpine (30 mg/kg) was administered, and the head electrode was connected to the recording wire. The EEG and video monitoring equipment were then turned on. The behavioral performance of each rat was observed and evaluated using the Racine scale. The seizures of rats in each group were closely observed; if seizures did not occur within 30 minutes, pilocarpine was injected intraperitoneally again (at half the initial dose), followed by 30 minutes of observation. If seizures did not occur, the model establishment was considered to have failed. Alternatively, if a grade V seizure was observed for 10 minutes, an intraperitoneal injection of diazepam (10 mg/kg) was administered, and changes in EEG signals and behavior were observed. If the seizure was unable to be terminated, the RSE model was considered to have been established successfully.
Next, to study the therapeutic effects of different doses of hydrogen on the RSE model, we used hydrogen-rich saline (HRS). Rats in the RSE + HRS group were treated intraperitoneally with different doses of HRS, whereas rats in the RSE + saline group were given a corresponding dose of normal saline 5 minutes after receiving diazepam (10 mg/kg). The EEG recording parameters were as follows: 30 mm/s, 2 mm = 50 V. The EEG recording started 5 minutes before the pilocarpine intraperitoneal injection and continued for 60 minutes of observation. The EEG data were processed using MATLAB software and analyzed via two different methods. We defined SE using the following EEG data: frequency ≥ 5 Hz, duration >30 s, and amplitude ≥ 3 times the baseline amplitude.
Western blot
After hippocampal extraction, plasma membrane protein was isolated using a Minute Plasma Membrane Protein Isolation and Cell Fractionation Kit (SM-005-4; Invent Biotechnologies, Inc., Plymouth, MN, USA). Equal amounts of protein samples were separated via sodium dodecyl sulfate–polyacrylamide gel electrophoresis and subsequently transferred to polyvinylidene fluoride membranes (ISEQ00010; Millipore, Billerica, MA, USA). After being blocked for 1 hour, the membranes were incubated overnight at 4°C with the appropriate primary antibodies—NR2B (ab28373, 1:500; Abcam, Cambridge, UK), phosphorylated (p)-NR2B (ab81271, 1:300; Abcam), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; CW0100 M, 1:2000; CoWin Biosciences, Taizhou, China)—followed by a 2-hour incubation at room temperature with secondary antibodies (goat anti-rabbit IgG, CW0103S, and goat anti-mouse IgG, CW0102S; both CoWin Biosciences). Immobilon Western Chemiluminescent HRP Substrate (P0018 M; Beyotime Biotechnology, Shanghai, China) was used to detect the bound antibodies. ImageJ software (NIH, Bethesda, MD, USA) was used to quantify band intensities. The NR2B and p-NR2B band intensities were normalized to the band intensity of GAPDH, which was used as a loading control.
In vitro experiments
Primary rat hippocampal neuronal cultures
Animals were purchased from the Animal Center of the Fourth Military Medical University. All experiments were performed on mixed primary hippocampal neuronal cultures that were prepared as described previously, with slight modifications. 19 First, hippocampal neurons were isolated from embryonic day 18 Sprague Dawley rat brains. The embryos were transferred and euthanized by decapitation in ice-cold dissection medium composed of Hank’s buffered salt solution. The cells were then seeded in Falcon 35-mm cell culture dishes or on poly-l-lysine-coated glass coverslips (0.05 mg/mL). The hippocampal cells were maintained at 37°C in an atmosphere of 5% CO2/95% air and cultured in neurobasal medium supplemented with 2% B27, 0.5 mM l-glutamine, 100 units/mL penicillin, and 100 g/mL streptomycin. The cells were maintained for 5 days before further analysis.
Mg2+-free condition
For the Mg2+-free condition, we prepared physiological bath recording solution (pBRS) without MgCl2 (low Mg2+), which was composed of (in mM): 145 NaCl, 2.5 KCl, 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 2 CaCl2, 0.002 glycine, and 10 glucose. The solution had a pH of 7.3, and the osmolarity was adjusted to 315 to 325 mOsm using sucrose. The neuronal cells were incubated with the solution for 2.5 hours to induce high-frequency epileptiform bursts (SE-like electrographic activity). The high-frequency epileptiform bursts continued until pBRS containing 1 mM MgCl2 was added to the culture. The pBRS without MgCl2 was used for the low-Mg2+ treatment condition, and controls were treated with pBRS containing 1 mM MgCl2.
Real-time fluorescence quantitative polymerase chain reaction (PCR)
Real-time fluorescence quantitative PCR was used to measure the mRNA expression of mixed lineage kinase domain-like protein (MLKL), caspase-3, and apoptosis regulator Bcl-2 at 3, 6, and 12 hours after treatment with Mg2+-free extracellular fluid. A real-time fluorescence quantitative kit was used (Takara Bio, Kusatsu, Japan) according to the manufacturer’s instructions. The primers used are displayed in Table 1.
Primers used in the polymerase chain reaction experiments

Statistical analysis
Data were analyzed using IBM SPSS Statistics for Windows, version 20.0 (IBM Corp., Armonk, NY, USA), and all experimental results are displayed as the mean ± standard error. All experiments were repeated three independent times. Student’s t-test was used to determine differences between two groups of data, whereas one-way analysis of variance (ANOVA) was used to compare data among multiple groups. Repeated measurements were tested using two-way ANOVA. For all tests, P < 0.05 was considered to indicate a significant difference.
Results
Benzodiazepine-resistant SE model
In the lithium-pilocarpine epilepsy model, previous findings suggest that when the seizure severity of an experimental animal reaches or exceeds grade V and lasts for 10 minutes, the animal begins to become resistant to diazepam and develops RSE. 20 On the basis of these findings, in the rat pilocarpine epilepsy model, we administered diazepam after the severity of epileptic seizures reached grade V, and maintained the dose for 10 minutes. To determine whether this model is appropriate for basic research on RSE, we then observed the therapeutic effects of benzodiazepines through behavioral tests and EEG. SE began to appear in rats approximately 18 ± 5.6 minutes after pilocarpine administration (n = 10). At 10 minutes after diazepam treatment, although EEG seizure intensity decreased and behavior improved, epileptic seizures were not effectively terminated (Figure 1). We therefore believe that this model can be used to simulate RSE for basic research.

Epileptic seizures reached grade V and remained responsive to benzodiazepines after 10 minutes. (a) Diazepam was given 10 minutes after the behavioral score reached or exceeded grade V, and the amplitude of the electroencephalography signal decreased; however, the seizure was not terminated (n = 10) and (b) behavioral scores of the model rats decreased after treatment (n = 10).
HRS treatment reduces the intensity of EEG attacks in RSE rats
After the model was successfully established, the animals were randomly divided into four groups (n = 6 per group). Animals in each group were treated with different doses of HRS (5, 10, 20, or 30 mL/kg) and observed. HRS at doses of 10 mL/kg and under produced therapeutic effects but did not reduce the amplitudes of EEG waveforms in rats (Figure 2a, b); however, HRS at doses of 20 and 30 mL/kg had similar therapeutic effects and were able to reduce EEG waveform amplitudes (P < 0.05) (Figure 2c, d).

Effects of different doses of hydrogen-rich saline (HRS) on electroencephalography (EEG) attack intensity in refractory status epilepticus (RSE) model rats. (a, b) After diazepam treatment, the EEG amplitude decreased, but the attack was not terminated. Treatment with 5 or 10 mL/kg HRS had no clear effect on EEG amplitude in rats (n = 6) and (c, d) treatment with HRS at 20 and 30 mL/kg further reduced EEG amplitude in rats after diazepam treatment (n = 6).
Next, we studied the therapeutic effects of HRS in more detail. At a dose of 30 mL/kg, the amount of liquid that needed to be injected intraperitoneally was high, which may affect the behavior of the RSE rats. We therefore selected 20 mL/kg as the therapeutic HRS dose. Over time, the intensity of EEG seizures began to increase in the rats (P < 0.05) (Figure 3). When the epileptic seizures reached intractable SE and the rats were treated with diazepam (10 mg/kg) for 10 minutes, both the EEG amplitude and power began to decrease. After the intraperitoneal injection of HRS, the EEG amplitude and power decreased further; this was followed by a loss of efficacy and a gradual increase in EEG amplitude and power (all P < 0.05). In the saline control group, the EEG amplitude and power did not change significantly after the saline injection but gradually increased over time.

Therapeutic effects of hydrogen-rich saline (HRS) on electroencephalography (EEG) amplitudes in refractory status epilepticus (RSE) model rats. (a) Saline treatment had no effects on EEG amplitude, and the EEG attack intensity gradually increased over time (n = 6) and (b) Rats were treated with 20 mL/kg HRS during continuous EEG recording. After HRS treatment, EEG attack intensity decreased before again increasing over time (n = 6).
HRS treatment does not improve the behavior of RSE rats
We further observed the behavioral effects of HRS treatment in rats. Pilocarpine was injected intraperitoneally to induce epileptic seizures. When the severity reached grade V for 10 minutes, diazepam was injected intraperitoneally for intervention. After 5 minutes, the animals were treated with the same dose of either HRS or normal saline, and behavioral changes were scored in the two groups of rats using the Racine scale to evaluate epilepsy severity at various time points after treatment intervention. Although SE was not terminated after treatment, the behavioral scores decreased to approximately 2 points in both groups of rats. Subsequently, the behavioral scores of the rats in both groups gradually increased over time (P < 0.05). There were no significant differences in behavioral scores between the HRS and saline groups (Figure 4).
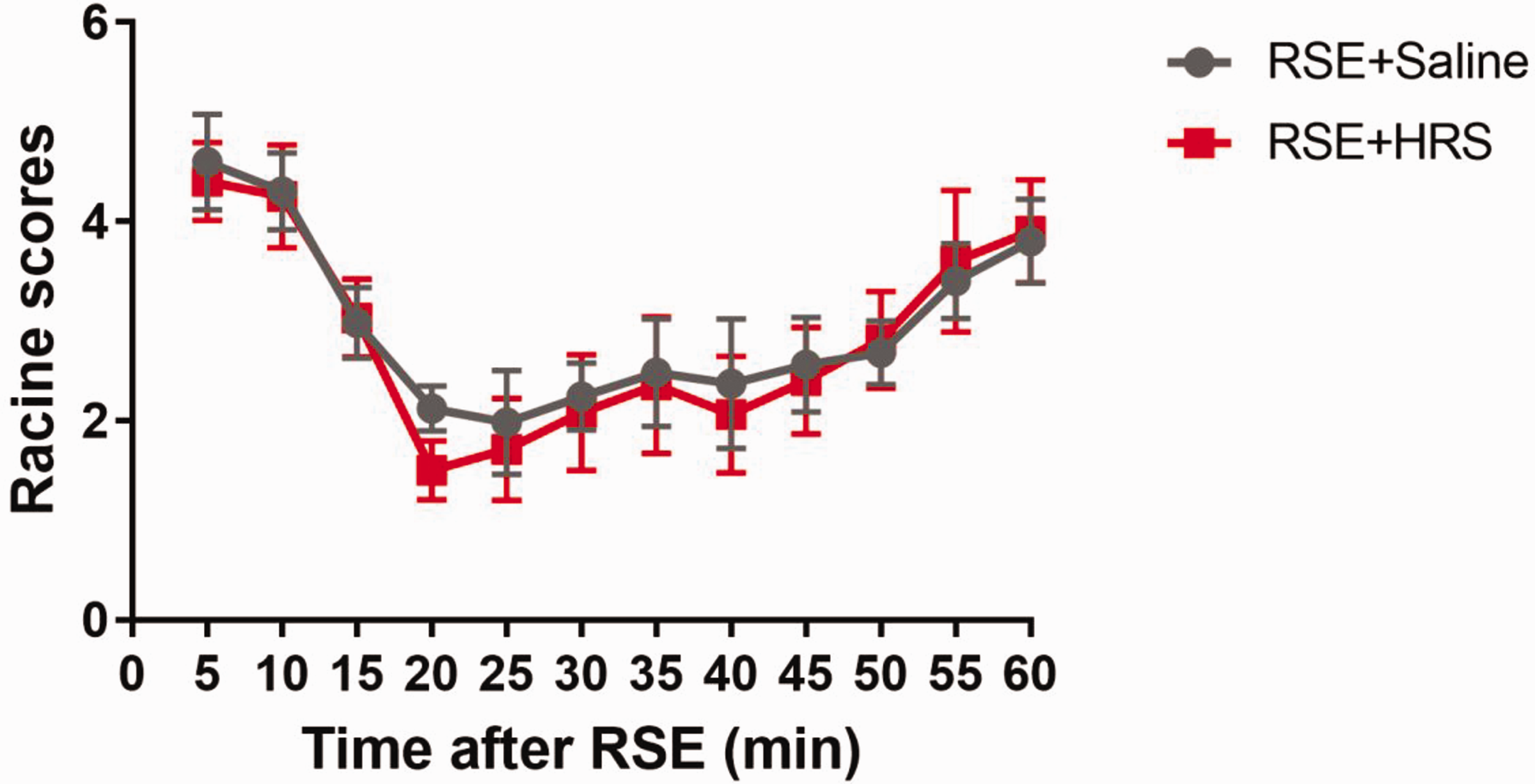
Effects of hydrogen-rich saline (HRS) treatment on the behavioral scores of refractory status epilepticus (RSE) model rats. Diazepam was given 10 minutes after RSE, and was followed by HRS or normal saline for 15 minutes. Although Racine scores did not differ between the two groups at any time point, the scores of both groups decreased after diazepam treatment before gradually increasing over time (n = 6). Data are shown as the mean ± standard error.
HRS treatment reduces NR2B phosphorylation on the cell membrane
To assess the therapeutic effects of HRS in RSE rats, we focused on the NR2B subunit, which is a functional regulatory subunit of the NMDA receptor.15,16 The NR2B subunit has multiple phosphorylation regulatory sites that can regulate the functional state of receptor channels, including receptor transport and expression. When the NR2B subunit is phosphorylated, the NMDA receptor-mediated current is enhanced. In the present study, we therefore treated RSE rats with HRS and observed changes in NR2B expression on hippocampal cell membranes. Hippocampal cell membrane NR2B expression gradually increased over time after RSE, and the levels of NR2B expression at 10, 15, and 30 minutes after RSE were greater than those in the control group (all P < 0.05) (Figure 5a, b). There were no significant differences in membrane NR2B expression at any time point between the RSE + saline and RSE + HRS groups (Figure 5c, d).15,16

Effects of hydrogen-rich saline (HRS) treatment on hippocampal cell membrane N-methyl-D-aspartate receptor subtype 2B (NR2B) expression. (a) Membrane NR2B expression was measured by western blot at different time points after refractory status epilepticus (RSE) induction (n = 3). (c) Membrane NR2B expression at different time points after RSE induction in the RSE + saline and RSE + HRS groups and (b, d) relative protein levels in each group (n = 3). Data are displayed as the mean ± standard error, *P < 0.05.
We further studied NR2B phosphorylation on the hippocampal cell membrane; the levels of p-NR2B also gradually increased over time (P < 0.05). Compared with the control group, p-NR2B levels were higher at 10, 15, and 30 minutes after RSE (all P < 0.05) (Figure 6a, b). Additionally, p-NR2B levels were significantly lower in the RSE + HRS group than in the RSE + saline group (P < 0.05) (Figure 6c, d). These results indicate that HRS can regulate the phosphorylation of NR2B. It has been reported that a decrease in NR2B phosphorylation can inhibit NMDA receptor function and reduce nerve excitability. 21–23 However, further experiments are needed to determine whether hydrogen regulates NMDA receptor function through this pathway and subsequently produces a therapeutic effect on RSE in rats.

Effects of hydrogen-rich saline (HRS) treatment on hippocampal cell membrane N-methyl-D-aspartate receptor subtype 2B (NR2B) phosphorylation. (a) NR2B phosphorylation was measured by western blot at different time points after refractory status epilepticus (RSE) induction (n = 3). (c) NR2B phosphorylation in the RSE + saline and RSE + HRS groups at different time points after RSE induction and (b, d) relative protein levels in each group (n = 3). Data are displayed as the mean ± standard error, *P < 0.05.
HRS treatment reduces oxidative stress in the SE model
To further explore the protective effects of HRS on hippocampal apoptosis and necroptosis in rats with RSE, we used primary neuronal cultures to establish an SE model and studied the related mechanisms in vitro. First, we used real-time fluorescence quantitative PCR to measure the mRNA expression of MLKL, caspase-3, and Bcl-2 at 3, 6, and 12 hours after treatment. Three hours after the neurons were treated with magnesium-free extracellular fluid to establish the SE model, caspase-3, MLKL, and Bcl-2 mRNA expression levels did not differ from those in the control group (treated with normal extracellular fluid) (Figure 7). At 6 hours after SE model establishment, caspase-3, MLKL, and Bcl-2 mRNA expression levels peaked in the cells (Figure 7b, c, d); these levels were significantly greater than those in the control group (all P < 0.05). At 12 hours after SE model establishment, caspase-3, MLKL, and Bcl-2 mRNA expression levels were significantly lower than those at 6 hours but remained higher than those in the normal control group (all P < 0.05). In addition, the number of surviving neurons in each group at the 24-hour time point supported the PCR results. The SE group had the fewest surviving neurons, and HRS treatment partially increased the number of surviving neurons (Figure 7a, e).

Effects of hydrogen-rich saline (HRS) treatment on the death of primary neurons cultured in magnesium-deficient medium. (a) Survival of neurons in each group 24 hours after status epilepticus (SE) model induction. (b, c, d) Real-time fluorescence quantitative polymerase chain reaction was used to detect the mRNA expression of caspase-3, mixed lineage kinase domain-like protein (MLKL), and apoptosis regulator Bcl-2; expression peaked at 6 hours and was greater at 6 and 12 hours after SE model induction compared with the control group and (e) the number of surviving neurons in each group in one field of view (200×) 24 hours after SE model induction. Data are displayed as the mean ± standard error, *P < 0.05.
The aforementioned results indicate that the in vitro SE model partly imitates the cell damage observed in vivo. We therefore explored the therapeutic mechanisms of HRS using this model. On the basis of the results of the previous experiment, we chose 6 hours after SE for all subsequent investigations. Changes in Bcl-2 and superoxide dismutase, mitochondrial (SOD2) expression in response to HRS treatment were detected using real-time fluorescence quantitative PCR. The mRNA expression of Bcl-2 was higher in the SE + saline group than in the control group, and HRS treatment significantly increased Bcl-2 expression (all P < 0.05) (Figure 8a). However, the mRNA expression of SOD2—an important antioxidative stress enzyme in vivo—was significantly lower in the SE + saline group than in the other three groups, including the SE + HRS group (all P < 0.05) (Figure 8b); this finding suggests that HRS may reduce oxidative stress by increasing SOD2 expression.

Effect of hydrogen-rich saline (HRS) treatment on apoptosis regulator Bcl-2 and superoxide dismutase, mitochondrial (SOD2) expression in cultured neurons. (a) Bcl-2 mRNA expression in each group was measured 6 hours after status epilepticus (SE) model induction and (b) effects of HRS treatment on SOD2 mRNA expression 6 hours after SE model induction. Data are displayed as the mean ± standard error, *P < 0.05.
Discussion
With the continuation of epileptic seizures, the therapeutic effects of drugs—especially benzodiazepines—gradually decrease; this highlights the potential value of glutamate receptor antagonists in the treatment of RSE. In particular, therapies targeting NMDA receptors may be an option for improving the current situation of epilepsy treatment. In the present study, HRS treatment did not improve the behavioral performance of RSE rats, but did reduce EEG amplitude. In addition, HRS treatment regulated the phosphorylation of the NR2B subunit of NMDA receptors, suggesting that the therapeutic effects of HRS may be mediated through this pathway.
At present, RSE treatments do not effectively reduce the mortality rate or improve prognosis. There is therefore an urgent need to screen suitable RSE animal models for further study. After carefully analyzing various existing epilepsy animal models, we noted that the lithium-pilocarpine model is resistant to benzodiazepines after the behavioral score is maintained at grade V for 10 minutes; 20 this model thus has potential as a benzodiazepine-resistant RSE animal model. We evaluated the model in detail using behavioral and EEG data. When a seizure severity of grade V was sustained for 10 minutes, we administered an intraperitoneal injection of diazepam (10 mg/kg) to the rats. Although diazepam did not terminate the seizures, both Racine scores and EEG amplitudes were reduced. Together, these findings suggest that these model rats are resistant to benzodiazepines at this time. We therefore used this model to observe the therapeutic effects of hydrogen.
There is currently no doubt that hydrogen can be used to treat or prevent various diseases. For example, Ohsawa et al. first reported that hydrogen can significantly reduce oxidative stress and exert neuroprotective effects in an inhalation-induced rat model of focal cerebral ischemia–reperfusion injury. 11 Further studies later confirmed the therapeutic effects of hydrogen on cerebral ischemia through multiple injections of HRS.24,25 Ogawa et al. and Xu et al. reported that one sufficient dose of HRS can be used to treat hearing loss caused by ischemia and carbon monoxide-related toxic encephalopathy.26,27 In addition, orally administered hydrogen-rich water may also have beneficial effects in patients with Parkinson's disease or hepatitis B.28,29 Together, these results indicate that hydrogen can exert therapeutic effects when administered at various doses and for various durations via various routes.11,24–27,29–31
In the current study, after comprehensive consideration, we chose an intraperitoneal injection of HRS as the most feasible method of administering hydrogen to study the effects of HRS on seizures in RSE rats. We first screened therapeutic doses and found that HRS at both 20 and 30 mL/kg was able to reduce EEG amplitudes in RSE rats. Given that a dose of 30 mL/kg requires additional HRS to be injected without a substantial increase in efficacy, 20 mL/kg was ultimately selected as the optimal therapeutic dose. We subsequently conducted a further study on the therapeutic effects of HRS and found that EEG amplitudes in model rats decreased after HRS treatment and then increased again over time. This finding is consistent with the results reported by Ono et al. 14 In addition, our behavioral observations revealed no significant differences in behavioral performance between RSE rats after HRS treatment and rats in the normal group after saline treatment. This finding suggests that the therapeutic effects of HRS may not be sufficient to improve the behavioral performance of model rats. 14
There is an important transition process in epilepsy from a seizure state to a post-seizure state. The failure of this mechanism to terminate epilepsy leads to a prolonged seizure duration, which is partly the result of reduced drug efficacy caused by GABAAR internalization. The internalization of GABAAR on the synaptic membrane can lead to tolerance to benzodiazepines during SE in model rats, whereas the aggregation of NMDA receptors on the membrane gradually increases the efficacy of NMDA receptor antagonists and other NMDA receptor-targeting drugs in SE. In the electroconvulsive model, administration of a GABAAR-positive allosteric agent 15 minutes after the seizure can control 66% of seizures in rats, but at 1 hour it can control only 25% of seizures. 32 In contrast, blocking NMDA receptors with MK-801 or ketamine can quickly terminate seizures. 33 In addition, acute changes in other receptors caused by epileptic seizures also promote the persistence of epileptic seizures; for example, decreased presynaptic adenosine A1 receptor expression and loss of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor GluA2 subunit.20,34,35 AMPA receptors lacking the GluA2 subunit exhibit calcium permeability, which can lead to calcium overload and eventually neuronal death. These results suggest that drugs targeting NMDA receptors may offer new possibilities for the treatment of RSE.20,32–35
Hydrogen has many known functions, including anti-inflammatory, antiapoptotic, and antiallergic functions, and can promote energy metabolism. 11 However, attributing the therapeutic effects of hydrogen only to well-known antioxidant functions is unreasonable because the antioxidant effect of hydrogen is less than 1/100 of that of vitamins C and E; 36 more importantly, hydrogen molecules can directly regulate the function of NMDA through NR2B to play a therapeutic role. We therefore wondered: can HRS also regulate NR2B?
To further explore the effects of HRS on NR2B, we observed the levels of NR2B expression and phosphorylation on the hippocampal cell membrane. Hippocampal cell membrane NR2B expression gradually increased after epilepsy, which is consistent with the results of previous research.37–41 After HRS treatment, hippocampal cell membrane NR2B expression was not significantly different between rats in the treatment and control groups, suggesting that HRS treatment cannot change the pathological process of NR2B aggregation on the hippocampal cell membrane during SE. Therefore, HRS does not affect NR2B expression on the hippocampal cell membrane in RSE. Furthermore, we observed changes in NR2B phosphorylation levels on the hippocampal cell membrane; these levels gradually increased over time after epilepsy but were reduced by HRS. Together, these results suggest that HRS may play a therapeutic role by affecting NMDA function via reduced phosphorylated NR2B levels; however, this hypothesis requires further confirmation.
The preset findings indicate that hydrogen can reduce EEG intensity in RSE rats. Moreover, the functions of hydrogen in resisting oxidative stress and reducing inflammation cannot be ignored. As such, can the decreases in EEG intensity and the antioxidative stress and anti-inflammatory effects resulting from HRS treatment reduce the degree of neuronal death caused by RSE? To detect the effects of HRS on the expression of antioxidant enzymes and antiapoptotic molecules in primary cultured neurons, we simulated pathological changes in vivo using a magnesium-free cell model. HRS increased both SOD2 and Bcl-2 expression, which suggests that HRS can play a therapeutic role by increasing antioxidant enzyme expression and subsequently reducing the oxidative stress damage induced by RSE.
In clinical research, benzodiazepines are first-line drugs for the treatment of epilepsy. Benzodiazepines are very effective at terminating early seizures; however, over time, the ability of these drugs to terminate seizures gradually decreases.7,42 In some animal models, NMDA receptor antagonists alone cannot terminate benzodiazepine-resistant seizures. Nonetheless, when these drugs are used in combination with benzodiazepines, seizures can be terminated.43–45 This indicates that NMDA receptor antagonists and benzodiazepines have synergistic effects. Hydrogen, which can directly act on NMDA receptors, may therefore play an increasingly important role in RSE treatment in the future because of its excellent characteristics.
The present findings indicate that hydrogen may reduce seizure intensity in RSE model rats by acting on NMDA receptors, and may also exert neuroprotective effects via antioxidative stress effects and reducing inflammation, among other mechanisms. However, there are some limitations to our study. For example, we did not analyze the specific pathways involved in oxidative stress that result in cell death, and we did not evaluate how NMDA receptors might decrease seizure intensity. Further exploration of the underlying mechanisms is therefore needed.
Conclusions
Hydrogen may reduce seizure intensity in RSE model rats by acting on NMDA receptors. It may also exert neuroprotective effects by reducing oxidative stress and inflammation, among other mechanisms. Hydrogen is advantageous because it is safe and can easily diffuse across the blood–brain barrier; it may thus be used in combination with benzodiazepines to treat SE in the future to improve patient prognosis.
Footnotes
Acknowledgements
We thank AJE for providing language editing services.
Author contributions
All authors had full access to all data in the study and take responsibility for its integrity and the accuracy of the data analysis. Conceptualization: XC and WJ; methodology: RJ, RZ, and GZ; investigation: RJ, RZ, and GZ; formal analysis: RJ; writing – original draft: RJ and GZ; writing – review & editing: TL, XC, and WJ; visualization: RJ and WJ; supervision: XC; funding acquisition: RJ.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
Support for this work came from the Shaanxi Natural Science Foundation (grant Nos: 2022JQ-754).