
Abstract
Strokes are the leading cause of death in most regions of the world. Epoxidase inhibitors include the drug aspirin (acetylsalicylic acid). Aspirin is widely used as first-line treatment for the prevention of cardiovascular and cerebrovascular diseases in at-risk patients. However, patients using conventional doses of aspirin can still develop ischaemic cardiovascular and cerebrovascular diseases, a phenomenon known as aspirin resistance. The occurrence of aspirin resistance hinders the prevention and treatment of ischaemic cardiovascular and cerebrovascular diseases. There are many factors affecting aspirin resistance, such as sex, drug dose, metabolic disease, genetic polymorphisms, drug interactions and pharmacokinetics. Genetic polymorphism refers to the simultaneous and frequent presence of two or more discontinuous variants or genotypes or alleles in a population of organisms. Platelets contain a large number of highly polymorphic transmembrane glycoprotein receptors encoded by two or more isomeric alleles. Changes in gene polymorphisms in various pathways during platelet aggregation can lead to aspirin resistance. This narrative review describes the gene polymorphisms that have been demonstrated to be significantly associated with aspirin resistance. Research on the mechanisms of aspirin resistance and increased knowledge should provide accurate drug guidance in individuals that require first-line antiplatelet therapy.
Keywords
Introduction
Platelets play an important role in the pathogenesis of ischaemic cardiovascular and cerebrovascular diseases and adhere to ruptured unstable atherosclerotic plaques. As an antiplatelet drug, aspirin is widely used in the prevention of cardiovascular and cerebrovascular diseases at all levels. However, some patients still have ischaemic events after using the conventional dose of aspirin.1,2 At present, the widely recognized definition of resistance to antiplatelet drugs refers to platelet aggregation not being completely inhibited as determined by platelet function being detected after the application of antiplatelet drugs. 3 Due to the use of different methods of measuring platelet function, variable study populations and different criteria for the definition of platelet resistance, the incidence of antiplatelet drug resistance reported by reported studies varies greatly. For example, based on the existing literature, the incidence of aspirin resistance in the population ranges from 0.4% to 83.3%. 4 The exact mechanism of antiplatelet drug resistance has not been fully elucidated. Studies have shown that gene polymorphism is one of the factors affecting antiplatelet drug resistance and this will be discussed further in this narrative review.
For this narrative review, articles published before 1 January 2022 were searched in the PubMed®, Web of Science and Chinese (China National Knowledge Network, Wanfang Data and Chinese Technical journals) databases using the following search terms: (“aspirin resistance” or “aspirin resistance”) and (“genetic polymorphism” or “polymorphism”). This review includes randomized or quasi-randomized prospective controlled clinical trials, reports, guidelines and letters to the editor, which were published in English and Chinese. This narrative review discusses the relevant studies, focusing on the relationship between aspirin resistance and genetic polymorphisms, as well as potential drug development targets, with the aim of providing recommendations for clinicians.
Overview of cyclooxygenase inhibitors and aspirin
Cyclooxygenase inhibitors
Cyclooxygenase (COX), also known as prostaglandin oxidase reductase, is an enzyme with dual functions. It not only functions as a cyclooxygenase but it also has catalase activity. It can also catalyse the conversion of arachidonic acid into prostaglandin. It can regulate female reproductive function, childbirth, platelet aggregation and cardiovascular system balance. 5 Cyclooxygenase has two isoenzymes, COX-1 and COX-2. COX-1 is structural and mainly expressed in tissues such as kidney, blood vessels and stomach; and it has functions of regulating platelet aggregation, protecting gastrointestinal mucosa, regulating renal blood flow distribution and regulating peripheral vascular resistance. 6 The activity of COX-2 in normal tissue cells is extremely low, but when the cells are stimulated by inflammation, its expression level in inflammatory cells can be increased to 10–80 times the normal level, leading to inflammatory reactions and tissue damage, so COX-2 is known as an inducible enzyme. When the body is stimulated by various physical and chemical factors, phospholipase A2 can be activated, and the enzyme hydrolyses cell membrane phospholipids to generate arachidonic acid, which is then catalysed by COX-2 to produce prostaglandins. 7 Cyclooxygenase inhibitors are compounds that inhibit cyclooxygenase, which include non-specific cyclooxygenase inhibitors that inhibit both COX-1 and COX-2, such as aspirin; and specific COX-2 inhibitors, such as celecoxib and celecoxib.
Overview of aspirin
In 1853, Gerhardt first synthesized acetylsalicylic acid from salicylic acid and acetic acid. Later, the German chemist Felix Hoffmann used the synthetic aspirin to treat rheumatoid arthritis, which proved to be effective. Therefore, aspirin was gradually recognized and popularized for clinical treatment by Heinrich Dreser in 1899. Acetylsalicylic acid was officially named aspirin and its chemical formula is C9H8O4 (Figure 1), with a relative mass of 180.16.8,9
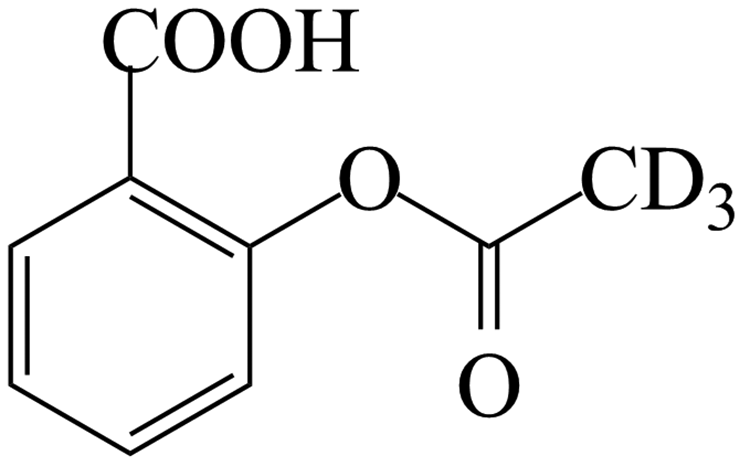
Molecular structure of aspirin.
Antiplatelet aggregation mechanism of aspirin
In terms of antiplatelet aggregation, different concentrations of aspirin have different mechanisms. 10 Low concentrations of aspirin cannot acetylate prostaglandin synthetase, resulting in the inhibition of platelet cyclase. After the inhibition of this enzyme, the production levels of thromboxane A2 are significantly decreased, thus affecting platelet aggregation and thrombosis and playing an antiplatelet aggregation role. High concentrations of aspirin directly inhibit prostaglandin production in blood vessel walls, reducing the ability to antagonize thromboxane A2, which has a strong platelet-inducing effect and thus antithrombotic formation (Figure 2).

Mechanisms of action of aspirin in terms of reducing platelet aggregation. PGG2, prostaglandin G2; PGH2, prostaglandin H2; PG12, prostaglandin I2; TXA2, thromboxane A2; cAMP, cyclic adenosine monophosphate. The colour version of this figure is available at: http://imr.sagepub.com.
Aspirin resistance and gene polymorphisms
Aspirin has a wide range of effects, including antipyretic and analgesic effects, anti-inflammatory effects, antiplatelet aggregation, anti-tumour effects, among which antiplatelet aggregation is one of the most important. At present, aspirin has been listed as a cornerstone drug for the prevention and treatment of cardiovascular and cerebrovascular diseases. However, new vascular events still occur in some patients even after aspirin use, a phenomenon known as clinical aspirin resistance. 11 In addition, laboratory tests have found that in some patients aspirin does not inhibit thromboxane A2 production, which is called biochemical aspirin resistance. 12 At present, studies have shown that there are many reasons leading to aspirin resistance, such as poor drug compliance and insufficient drug dose, non-cyclooxygenase 1 mediated platelet activation, obesity and gene polymorphisms;13–16 of which gene polymorphisms are the focus of considerable research in aspirin resistance at present.17–20 Platelets contain a large number of highly polymorphic transmembrane glycoprotein receptors encoded by two or more isomeric alleles. Genetic polymorphism refers to the simultaneous and frequent presence of two or more discontinuous variants or genotypes or alleles in a population of organisms. Therefore, changes in gene polymorphisms in various pathways during platelet aggregation can lead to aspirin resistance. Studies have shown that the role of gene polymorphism in aspirin resistance is approximately 14–39%. 21 Currently, it has been found that glucose-6-phosphate isomerase a (GPIa) gene, glucose-6-phosphate isomerase b (GPIb) gene, cyclooxygenase (COX) genes (COX-1 and COX-2), platelet endothelial aggregation receptor 1 (PEAR1) gene and other gene polymorphisms are significantly related to aspirin resistance. These will be described in more detail below.
COX genes
The COX-1 and COX-2 genes play different roles in the mechanistic pathway of aspirin, so polymorphic changes of either of these two genes may cause aspirin resistance. The COX-1 gene is located on the 32–33.3 zone of the long arm of human chromosome 9; and it has a length of approximately 22 kilobases with a molecular structure consisting of 10 introns and 11 exons. 22 At present, the common mutation sites in the 5 terminal non-coding regions of the COX-1 gene are A-842G, 1676 T-C, G-1498A, A1201G, T1794C, G-10006A, A-918G and A-807G.23–28 The -842 A–G allele mutation was present in 60% of aspirin-resistant patients, while only 17% of non-aspirin-resistant patients had the mutation at the above site, suggesting that the A-842G allele mutation is closely related to aspirin resistance. 25 It was further found that the polymorphic changes at this gene locus could significantly increase the expression level of thrombin B2 and stimulate platelet aggregation, leading to an increased risk of thrombosis. 29 The 1676 A–G polymorphisms increase the risk of aspirin resistance in patients with coronary heart disease (odds ratio [OR] 1.82; 95% confidence interval [CI] 1.13, 2.92; P = 0.01). 30 In addition, other studies have shown that people with the −50 C–T gene mutation who take aspirin have a lower risk of developing colorectal polyps than those who do not take aspirin; and the mechanism might be that the gene polymorphism interferes with the genetic pharmacological effect of aspirin. 31 However, a study has also shown that the −50 C–T gene polymorphism is not associated with aspirin, which may be due to environmental factors influencing aspirin resistance. 32 A meta-analysis showed that the COX-1 gene was not significantly correlated with aspirin resistance, so the clinical application of COX-1 gene polymorphisms in relation to aspirin resistance is not currently used. 33 COX-2 gene polymorphisms can also cause aspirin resistance. The COX-2 gene is located on the 25.2–25.3 region of the long arm of human chromosome 1 and there are two GC and CC genotypes in the promoter sequence at −765. 34 Among them, the expression level of COX-2 mRNA in platelets and endothelial cells of patients with the CC genotype was significantly increased, leading to the increased production of COX-2 protein and thus aspirin resistance, suggesting that patients with −765CC polymorphism might have a higher risk of adverse vascular events. 35 However, a meta-analysis has shown that the −765 G–C polymorphism is not significantly correlated with vascular diseases, but this might related to the sample size and different patients that were included in the studies. 36
Platelet membrane glycoprotein genes
Platelet aggregation is dependent upon platelet activation, which is in turn dependent upon the binding of the glycoprotein Ia/IIa (GPIa/IIa) complex to collagen.37,38 The nucleotide sequence at the −807 site in the GPIa gene is significantly correlated with the number of GPIa/IIa molecules. 39 The number of GPIa/IIa molecules in patients with the TT and TC genotypes is significantly higher than that in those with the CC genotype, resulting in increased collagen binding ability, platelet activation and aggregation, and aspirin resistance. 39 In addition, the TT genotype can also lead to increased GPIa/IIa receptor density and thus aspirin resistance.40,41 A study found that the occurrence of non-fatal myocardial infarction was significantly correlated with the GPIa-807T-C allele. 42 The GPIa C807TT allele phenotype is generally associated with an increased risk of ischaemic cerebral infarction in the population, and thus GPIa gene polymorphism in platelet membranes is presumed to be a genetic predisposition factor for thrombotic disease. 39 However, the correlation between platelet glycoprotein gene polymorphism and aspirin resistance is still inconclusive and needs to be confirmed by further research data.
The final common pathway for platelet activation is via the GPIIb/IIIa receptor. Mutations in the genes encoding platelet GPIIb and GPIIIa can change the expression level and structure of GPIIb and GPIIIa, thus affecting platelet activation and aggregation. Changes in the second exon sequence of the GPIIIa gene can lead to the translation of leucine into proline. 43 A meta-analysis demonstrated that patients with stable coronary heart disease that have the above gene mutations have a significantly higher risk of acute ischaemic cardiovascular events than those without mutations, which may be due to the increased fibrinogen binding ability of GPIIb/IIIa with proline, resulting in increased platelet aggregation. 33 GPIIIa PlA2 allele carriers are more susceptible to aspirin resistance than the PlA1/PlA1 genotypes. 44 Patients with PlA2 alleles were found to have significantly increased restenosis rates after percutaneous coronary intervention (47% versus 38%; OR 1.42; 95% CI 1.09, 1.84), with PlA2 homozygous carriers at the highest risk, and this phenomenon was more pronounced in female patients. 45 However, some studies have also found that PlA2 gene polymorphism is negatively correlated with the occurrence of early stent thrombosis events. 46 A meta-analysis found that in patients with coronary heart disease, there was no significant difference in the incidence of laboratory aspirin resistance between PlA1/A1 genotype patients and PlA2 gene carriers (29.7% versus 28.3%; OR 0.94; 95% CI 0.73, 1.40). There was also no significant difference in adverse clinical outcomes between PlA2 carriers and PlA1/A1 genotype patients. 47 Therefore, currently, individualized antiplatelet therapy guided by PlA1/A2 gene testing for patients may not be clinically meaningful.
Platelet endothelial aggregation receptor 1 gene
Platelet endothelial aggregation receptor 1 is a type I cell surface receptor and the PEAR1 gene is composed of 23 exons and 22 introns. 48 Phosphorylation of this receptor can promote the activation of the glycoprotein IIb/IIIa fibrinogen receptor, promoting the activation and amplification of its signal, which leads to a large amount of platelet degranulation that causes an irreversible aggregation reaction between platelets, so it plays an important role in the formation of thrombus. 49 At present, most research is focussed on the rs12041331 and rs12566888 polymorphic sites.50,51
rs12041331 polymorphisms
The rs12041331 polymorphic site is considered to be the site most associated with platelet function, accounting for 15% of the total variation of platelet function phenotype. 52 The G allele increases the expression of the PEAR1 protein. 53 In patients treated with aspirin, the wild-type GG genotype had the highest expression, while the homozygous mutant AA genotype had the lowest expression. 54 The rs12041331 polymorphic site can significantly affect the non-COX-1-dependent platelet aggregation pathway and thus affect the antiplatelet aggregation effect of aspirin. 55 Patients with rs12041331 allele A had a relatively reduced platelet aggregation rate and the GG genotype was an independent risk factor for aspirin resistance. 56 However, some studies have obtained different results, demonstrating that patients carrying the A allele have a significantly higher platelet aggregation rate, which increases the risk of aspirin resistance and ischaemic events.57–59 The homozygous AA genotype of the rs12041331 polymorphism is associated with severe resistance to aspirin, which affects the prognosis of patients. 60
rs12566888 polymorphisms
Research has suggested that the rs12566888 polymorphic site has the strongest association with platelet reactivity after aspirin treatment, suggesting that it may be a valuable therapeutic target. 61 Compared with rs12041331, rs12566888 is associated with platelet aggregation phenotypes and may be associated with other PEAR1 genotypes, while rs12041331 has a relatively independent effect on platelet aggregation. 55
Other PEAR1-related gene polymorphisms
The secondary allele C of the rs2768759 polymorphic site of the PEAR1 gene may lead to an increased risk of repeat vascular events. 62 Other research has found that the C allele of rs11264580, the G allele of rs2644592, the T allele of rs3737224 and the T allele of rs41273215 were closely related to high platelet reactivity and were independent prognostic factors. 63 The homozygous TT genotype of rs57731889 was associated with low platelet reactivity and was an independent predictor of low platelet reactivity. 64 Research has have found that the T allele of the PEAR1 rs56260937 polymorphism is an independent predictor of vascular remodelling events. 65 Polymorphisms at different sites in the PEAR1 gene have different effects on platelet reactivity. 66 The mechanistic relationship between PEAR1 gene polymorphisms and platelet function needs to be further studied.
P2RY1 and P2RY12 genes
Purinergic receptor P2Y1 and purinergic receptor P2Y12 are G-protein coupled receptors on the surface of platelets. 67 Adenosine diphosphate (ADP) is released after vascular injury. 67 ADP binds to the P2Y1 receptor triggering an increase in calcium ion levels and promoting platelet aggregation. 67 ADP binds to the P2Y12 receptor, which blocks the action of adenylate cyclase, reduces the intracellular level of cyclic adenosine phosphate and accelerates platelet aggregation. 68 Research has shown that the presence of rs1065776 polymorphism in the P2RY1 gene attenuates the effect of aspirin in healthy volunteers. 69 The rs9859538 polymorphism in the P2RY1 gene was associated with residual platelet reactivity (RPR). 70 P2RY12 gene polymorphisms rs1491974, rs3732765, rs10513398 and rs10935841 were moderately correlated with RPR. 70 P2RY12 gene polymorphisms rs7615865, rs1388623, rs1388622, rs7634096 and rs7637803 were slightly correlated with RPR. 70 P2RY1 gene polymorphisms rs1439010, rs701265, rs2312265, rs1371097 and rs12497578 were moderately correlated with RPR. 70
Other genes
The expression product of multidrug resistance gene 1 (MDR1) is P-glycoprotein, which has adenosine triphosphate-dependent transmembrane transport activity so it can transport drugs outside the cell, allowing the cell to acquire drug resistance. 71 Research has demonstrated that people with the MDR1 TT genotype have a lower risk of aspirin resistance than those with CC and CT genotypes. 72 Thromboxane A2 is an important molecule in the process of platelet aggregation and thrombosis. 73 It can play a role only when it binds to the thromboxane A2 receptor. 74 The podocyte thromboxane A2 receptor gene is the gene that encodes the thromboxane A2 receptor. 75 A study found that about 81.8% of patients with the CC genotype had low aspirin responsiveness. 72 The probability of non-CC genotype patients developing aspirin hyporesponsiveness was approximately 62.4%. 72 In addition, studies have found that gene polymorphisms of the uridine diphosphate glucuronyl transferase and leukotriene C4 synthetase genes are also associated with aspirin resistance.76,77 The occurrence of aspirin resistance in patients with ischaemic cerebral infarction may be the result of the combined action of multiple gene polymorphisms. High-risk interactions between genotypes of COX-2 s1371097 and GPIIIa rs2317676 may be closely associated with aspirin resistance. 65 The interaction between COX-1 rs3842787 and COX-2 rs20417 genotypes was also positively correlated with aspirin resistance. 78 Therefore, multigene testing and analysis of gene interactions to guide individualized medication of patients can avoid severe ischaemic disease complications due to low drug response in patients.
It should be noted that the increase in studies investigating aspirin resistance has resulted in evidence that the correlation between gene polymorphisms and ischaemic events is not direct. For example, a Belgian study analysed nine single nucleotide polymorphisms in the PEAR1 gene and found no correlations between gene polymorphisms and ischaemic events. 79 Another analysis of 11 gene polymorphisms, including PEAR1, found that gene polymorphisms were associated with aspirin resistance in vitro, but not with ischaemic events. 80 Therefore, there is insufficient evidence to directly extrapolate the in vitro laboratory results to the clinical situation, because thrombus formation in vivo is an extremely complex process. The rational for using genetic polymorphisms to guide clinical antiplatelet therapy needs further investigation.
Conclusion
The antiplatelet effect of aspirin therapy shows considerable individual variation, which is one of the important reasons for the recurrence of cardiovascular and cerebrovascular diseases in some patients. Focusing research on the relationship between gene polymorphisms and aspirin resistance will provide in-depth knowledge of the different mechanisms involved, which will lead to the development of new drugs targeting aspirin resistance. Although many studies have been undertaken on the genetic factors of aspirin resistance in recent years, there is still no unified consensus on which genes and mechanisms result in functional changes in platelets. The extent to which genetic factors affect platelet aggregation after aspirin treatment remains unclear. More large sample multicentre prospective studies are required to clarify the exact relationship between these factors.