
Abstract
Objective
Respiratory syncytial virus (RSV) and respiratory adenovirus (ADV) are two common pathogens that cause acute respiratory tract infections in children. We aimed to develop a rapid method for detecting both pathogens simultaneously.
Methods
The recombinase polymerase isothermal amplification (RPA) method was combined with the CRISPR/Cas detection system. The assay’s specificity and sensitivity were explored by designing RPA primers and CRISPR RNAs (crRNAs) through multi-sequence comparisons, optimizing the reaction conditions, and using a fluorescent reading device. The consistency of the test results of 160 clinical pharyngeal swab samples was studied using quantitative polymerase chain reaction (qPCR) results as a comparative control.
Results
RSV and ADV could be detected at levels as low as 104 copies/mL and 103 copies/mL, respectively, within 50 minutes with no cross-reactivity with other similar pathogens. For the clinical samples, compared with the qPCR method, the sensitivities for RSV and ADV were 98.1% and 91.4%, respectively, and the detection specificities were both 100%. The Kappa values were greater than 0.95, suggesting a high degree of consistency.
Conclusion
This method for detecting RSV and ADV is rapid, sensitive, and specific. It can accurately detect mixed infections in a timely manner, making it suitable for use in areas with scarce healthcare resources.
Keywords
Introduction
Acute respiratory tract infections are common respiratory diseases in children that are usually caused by viruses. 1 Respiratory syncytial virus (RSV) and respiratory adenovirus (ADV) are common pathogens of pediatric respiratory tract infections. These can cause common cold symptoms, such as cough and runny nose, in mild cases, while serious complications, such as pneumonia and otitis media, can occur in severe cases.2–6 Currently, virus isolation and culture, immunology, polymerase chain reaction (PCR)-based molecular biology, and other methods are used worldwide for detecting RSV and ADV, all of which have certain limitations.7–12 According to the recommendations of the World Health Organization (WHO), the detection of pathogens should be rapid, accurate, sensitive, and specific and should not require specialized technicians or large instruments. 13 Therefore, it is particularly important to develop an assay for RSV and ADV that can meet the above detection requirements.
As a new nucleic acid amplification technology developed in recent years, isothermal amplification technology (IAT) mainly includes recombinase polymerase amplification (RPA), loop-mediated isothermal amplification (LAMP), crossing priming amplification (CPA), strand displacement amplification (SDA), nucleic acid sequence-based amplification (NASBA), rolling circle amplification (RCA), and helicase-dependent amplification (HDA).14–17 Among these methods, the amplification reaction of RPA is performed at 25°C to 42°C, with an optimal reaction temperature range of 37°C to 42°C. Additionally, RPA does not have a thermal denaturation step, which can support the exponential amplification of pathogen-derived nucleic acid in a sensitive, specific, and rapid manner. This results in lower equipment requirements that can allow the use of portable rapid nucleic acid detection. RPA has been widely used for detecting viruses, bacteria, and parasites,18–27 with good prospect for bedside detection and other applications. However, when IAT is applied alone, it is prone to false positive results because of its high sensitivity. 28
Clustered regularly interspaced short palindromic repeats (CRISPR) and their CRISPR-associated (Cas) proteins form an adaptive immune system guided by RNA. With the continuous development of the CRISPR/Cas system for genome editing applications, it has recently been found that some proteins in the system (class II Cas proteins of Cas12, Cas13, and Cas14) can specifically recognize the cleavage of a target nucleic acid molecule. They can also non-specifically cleave free single-stranded DNA (ssDNA) or single-stranded RNA (ssRNA) molecules. Taking advantage of this property, Cas proteins, target nucleic acids, and CRISPR RNA (crRNA) form a ternary complex that specifically cleaves target nucleic acids under the guidance of a crRNA, while non-specifically cleaving double-labeled ssDNA or ssRNA probes. The detection and typing of pathogens in a sample can therefore be accomplished by indicating the presence or absence of target nucleic acids through fluorescent signals,29–32 opening up a new era of molecular diagnostic technology. In recent years, CRISPR/Cas assays have been widely used for nucleic acid detection mostly in combination with other amplification technologies.32–37 It is characterized by high sensitivity, high specificity, fast speed, and low cost. However, at present, it is mainly applied for detecting a single pathogen, with less frequent use for detecting two or more pathogens.29,32,38,39
Currently, the related detection applications of multiple RPAs have also been conducted in the molecular testing field,40–42 but no application has yet been used for detecting mixed infections of acute respiratory pathogens. In this study, we established an RPA technology-based detection method that conducts simultaneous amplification of two pathogens followed by CRISPR. This approach combines the highly sensitive pre-amplification step with the highly specific detection step. This design addresses the shortcomings of the existing detection methods, such as their time-consuming nature and dependence on personnel and instrumentation. Additionally, this method can support the early and rapid detection of RSV and ADV in a pharyngeal swab sample, which provides a new direction for the clinical diagnosis of mixed infections of the two pathogens.
Materials and methods
Preparation of experimental materials and sample extraction
Between 2020 and 2022, throat swab samples from children with acute respiratory infections were collected from the clinical laboratory of Shenzhen Children’s Hospital. The presence of nine pathogens, including RSV, ADV, parainfluenza virus (PIV), rhinovirus (RhV), chlamydia pneumoniae (CP), mycoplasma pneumoniae (MP), influenza A virus (FluA), influenza B virus (FluB), and human neutrophilic pneumonia virus (hMPV), were all confirmed by laboratory testing methods. All patient details were de-identified. The throat swabs were divided into packages and stored at −80°C until use. Nucleic acids were extracted from the samples using the RNA extraction kit QIAamp Viral RNA Mini Kit (QIAGEN, Hilden, Germany). The samples were dispensed and stored at −20°C for short-term use, avoiding repeated freezing and thawing. Verbal informed consent was obtained from each child’s guardian when collecting the clinical samples, and approval for using those clinical samples for the study was obtained from the Medical Ethics Committee of Shenzhen Children’s Hospital. The reporting of this study conforms to STAND guidelines. 43
Design of RPA primers and crRNAs
The full genomic sequences of RSV and ADV were obtained from the NCBI genome database (https://www.ncbi.nlm.nih.gov/). MEGA7 software (https://www.megasoftware.net/) was used to identify sequences of the two pathogens with intraspecies conservation and interspecies variation. Finally, the RSV N protein gene and the ADV Hexon protein gene were found to be conserved sequences, and the target sequences were evaluated by Blast for parameters such as conservativeness, specificity, and GC content. For RPA primer design, 28 primer premier 5 (http://primerexplorer.jp/e/) was used (Supplementary Table 1). The primer sequences were sent to Shanghai Invitrogen (Shanghai, China) for synthesis. The protospacer adjacent motif (PAM) of Cas12a was searched for in the conserved region to design the respective universal crRNAs of RSV and ADV (Supplementary Table 1) and evaluated by CHOPCHOP (http://chopchop.cbu.uib.no//). The crRNAs were synthesized using Genscript (Nanjing, China).
RPA-CRISPR/Cas12a assay
The assay step consisted of two parts: RPA amplification and CRISPR/Cas detection. Following the initial amplification of the pathogen using RPA, this reaction was added to the CRISPR/Cas detection reaction (Figure 1). RPA-based isothermal amplification was performed using the Weifang Amp Future RT-RPA Liquid-Based Kit (Weifang, China), with a 50-μL amplification reaction. Briefly, 1.5 μL dNTPs (300 μM), 1.5 μL forward primer (600 nM), 1.5 μL reverse primer (600 nM), and 2 μL nucleic acid template were sequentially added to the tube, followed by 2.5 μL magnesium acetate (14 mM) to start the reaction. The total volume was brought up to 50 μL with nuclease-free water. For the negative control, nuclease-free water was added in place of the template. The PCR tubes were inverted five or six times to mix, centrifuged, and placed in a metal bath device set at 42°C for 30 minutes to conduct the amplifications. Subsequently, the amplification products were mixed with phenol-chloroform isopropanol 1:1 and centrifuged at 10,000 × g for 3 minutes. Then, the supernatant was aspirated and electrophoresed on a 1.5% agarose gel at 140 V for 30 minutes to determine the optimal primer pairs and concentrations of the two pathogens within the 400 to 800 nM concentration range. This was also done to optimize the reaction temperatures (from 37°C to 42°C) and reaction times (5 to 30 minutes) to identify the best reaction conditions. ImageJ software (National Institutes of Health, Bethesda, MD, USA) was used to analyze the relative quantitative values of each band on the agarose gels. The index values of the target length amplification product bands were compared.

Schematic diagram of the RPA-CRISPR/Cas12a assay detection steps.
The total volume of the CRISPR/Cas12a reaction was 20 μL, which consisted of 2 μL LbCas12a buffer, 0.24 μL crRNA (120 nM), 0.4 μL Cas12a (100 nM), 0.8 μL ssDNA (400 nM) (5′-6-FAM-TTATT-BHQ-1-3′), 44 and 2 μL RPA amplification product. The volume was brought up to 20 μL with nuclease-free free water. Two crRNAs were designed to cover the three common genotypes of ADV (B/C/E). Additionally, different concentrations of ssDNA (400 nM, 800 nM, and 1600 nM), crRNA (120 nM, 240 nM, and 360 nM), and Cas12a (100 nM, 200 nM, and 300 nM) were tested during the CRISPR/Cas detection process to determine the optimal combination to ensure that multiple genotypes were detected. The mixture was added into all-black 96-well microtiter plates, then 80 μL of nuclease-free water was added. The fluorescence signal intensity was detected using a BioTek (Winooski, VT, USA) Synergy H1 multifunctional enzyme labeling instrument (incident ray wavelength of 485 nm, emission wavelength of 535 nm) set at 37°C, and the detection was performed once every minute for 30 minutes. The reaction was set up with a negative control group (NC: nuclease-free water) and the experiment was repeated three times.
Specificity and sensitivity of the RPA-CRISPR/Cas12a assay
Nine common respiratory pathogens, including RSV and ADV, were collected and synchronized to assess the specificity of the RPA-CRISPR/Cas12a assay.
To verify its detection sensitivity, recombinant plasmids of the target amplification regions were constructed, the RSV and ADV RPA amplification products were ligated into the PUC57 vector, and the bacterial fluids were obtained by combining with Escherichia coli culture. Extraction was performed using the QIAGEN plasmid mini-extraction kit. Subsequently, the plasmid concentration (ng/μL) was determined using the nucleic acid quantifier qubit, then the molecular weight and concentration were calculated through the plasmid molecular weight calculation website (http://www.detaibio.com/sms2/dna_mw.htmL). Finally, the plasmid copy number was calculated. The reaction was repeated three times using a 102 to 107 copies/mL gradient dilution of plasmid DNA as a template and nuclease-free water as a negative control.
TaqMan qPCR assay
Next, qPCR primers and probes were designed for the RSV and ADV conserved regions. Relevant sequences: RSV forward primer (5′-GGCAAATATGGAAACATACGTGAA-3′), RSV reverse primer (5′-TCTTTTTCTAGGACATTGTAYTGAACAG-3′), and RSV probe (5′-FAM-CTGTGTATGTGGAGCCTTCGTGAAGCT-BHQ1-3′); ADV forward primers (5′-AGTCTTCGACGTGGTCAGAG-3′, 5′-CCGAGACGTACTTCAGCCT-3′), ADV reverse primers (5′-AGGTAGACGGCCTCGATGA-3′, 5′-GACCGGTCTGTGGTCACG-3′), and ADV probes (5′-FAM-TGCACCAGCCMCACCGCGG-TAMRA-3′, 5′-FAM-CACGGTGGCGCCTACGCACG-TAMRA-3′). The qPCR reactions were performed using the Vazyme AccurSTART U+ One Step RT-qPCR Probe Kit (glycerol-free) (Nanjing, China). The total reaction volume was 20 μL, which included forward primer (10 μM), reverse primer (10 μM), probe (10 μM), nucleic acid templates, and nuclease-free water. The qPCR assays were performed using a LightCycler® 480 Instrument II (Roche Diagnostics, Indianapolis, IN, USA) with the following cycling conditions: 37°C for 2 minutes for decontamination, 50°C for 15 minutes for reverse transcription, 95°C for 5 minutes for denaturation, then 40 cycles of 95°C for 10 nM s and 55°C for 30 s, followed by a 10-s cool-down at 37°C to detect FAM fluorescence signals. A sample was considered positive when the average cycle threshold (CT) value was <37 and considered negative when the average CT value was ≥37. Positive, negative, and blank controls were implemented, which respectively used known pathogen nucleic acid, a different pathogen nucleic acid, and nuclease-free water as a template. The experimental results were considered true and reliable when the positive control was positive and both the negative and blank controls were negative.
Consistency testing of clinical throat swab samples
To validate the utility of the established RPA-CRISPR/Cas12a method for clinical sample testing, 160 throat swab samples from children with respiratory tract infection symptoms were collected. After nucleic acid extraction, the samples were analyzed using the RPA-CRISPR/Cas12a assay in parallel with the qPCR method. Each test was repeated three times and inter-method concordance comparisons were performed using Cohen’s Kappa analysis.
Statistical analysis
To more accurately evaluate the performance of the test, we used the prevalence of the two pathogens with the test to estimate the number of patients and non-patients in the sample collection.
The calculation formula is as follows:


The approximate prevalence rates of RSV (20%) and ADV (14%) were substituted into the sample calculation formula.45,46 The estimated numbers of persons infected (positive) and non-infected (negative) with RSV were 43 and 11, respectively, while there were 62 patients with ADV (positive) and eight patients without ADV (negative).
When the test results of both the standard and diagnostic methods were positive, it was called a “True Positive”. When both results were negative, it was called a “True Negative”.
When the standard method result was positive but the diagnostic method result was negative, it was called a “False Negative”. When the standard method result was negative but the diagnostic method result was positive, it was called a “False Positive”.
Specificity (%) = True Positive/(True Positive + False Negative) × 100%
Sensitivity (%) = True Negative/(True Negative + False Positive) × 100%
The 95% CI calculations for sensitivity and specificity were conducted with http://vassarstats.net/clin1.html.
Data are represented as the mean ± standard deviation (SD), n = 3. The independent samples t-test was used to compare two groups, with analysis performed using GraphPad Prism 8 (GraphPad Software, Inc., La Jolla, CA, USA). For statistical significance, * indicates P < 0.05, ** indicates P < 0.01, and *** indicates P < 0.001, while ns indicates no statistically significant difference. Kappa values were analyzed using SPSS data analysis software version 26 (IBM, Armonk, NY, USA).
Results
RPA primer screening and optimization
Two pairs of primers were designed for the highly conserved RSV N gene sequence for RPA and product purification, with the final results verified by 1.5% agarose gel electrophoresis. Both pairs of primers showed bands of amplified products of target length. However, non-specific bands were observed in the RSV-F1/R1 primer pair amplification products, while better amplification was achieved using the RSV-F2/R2 primer pair with no primer dimerization or non-specific amplification (Figure 2a). All the four pairs of RPA primers showed bands of amplified product with target length. The agarose gel electrophoresis results suggested that all four RPA primer pairs produced amplification products of the same target length. Among these, the ADV-F3/R3 primer pair amplification product had a non-specific band, while the ADV-F4/R4 primer pair had a better amplification effect. The ADV-F4/R4 primer pair was also more effective compared with ADV-F1/R1 and ADV-F2/R2, with no primer dimerization or non-specific amplification (Figure 2b).
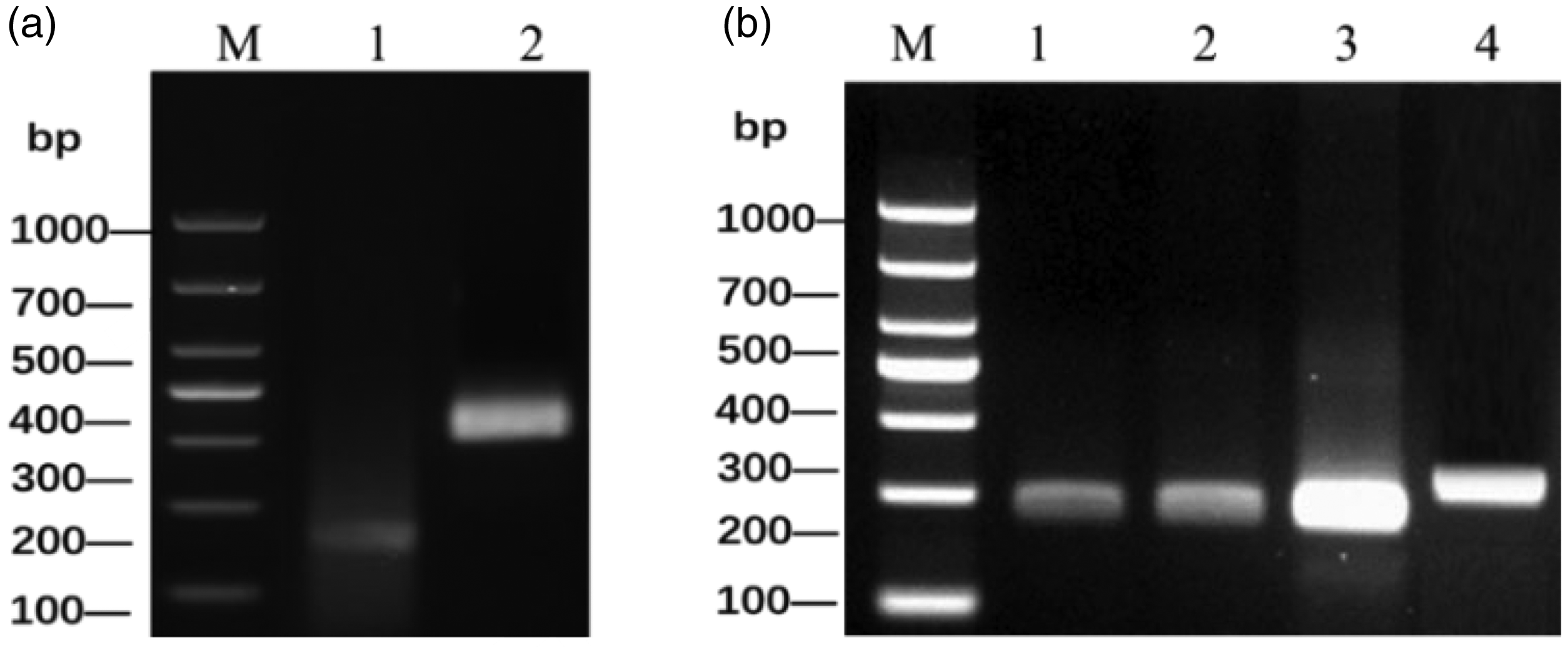
The screening procedure for the designed RSV and ADV primer pairs using agarose gel electrophoresis. 42 (a) M: DL 1000 DNA Marker; 1: RSV-F1/R1 primer pair; 2: RSV-F2/R2 primer pair and (b) M: DL 1000 DNA Marker; 1–4: ADV-F1/R1–ADV-F4/R4. RSV, respiratory syncytial virus; ADV, respiratory adenovirus.
After determining the respective RPA primers for RSV and ADV, primers for both pathogens were added to the RPA reaction for double amplification. The corresponding primer concentrations were set to 400 nM/400 nM, 400 nM/600 nM, and 400 nM/800 nM for RSV and ADV, respectively. The 1.5% agarose gel electrophoresis analysis showed that target length amplification products appeared with different primer ratios. Ultimately, the 400 nM RSV and 600 nM ADV primer concentration combination was selected by ImageJ software analysis (Figure 3a). Subsequently, the dual RPA reaction temperature range was set from 37°C to 42°C, but the amplification effect did not change significantly in this range. The amplification reaction temperature was finally selected as 40°C using ImageJ analysis (Figure 3b). The RPA reaction time range was set from 5 to 30 minutes. The amplified product band brightness was enhanced between 5 and 15 minutes, but did not change significantly from 20 to 30 minutes. ImageJ-based analysis was used to selected 20 minutes as the final detection time (Figure 3c).

Optimization of the RPA conditions for RSV and ADV, including the primer concentration ratio, reaction temperature, and reaction time. (a) M: DL 1000 DNA Marker; 1–3: respective RSV and ADV primer concentration combinations: 1: 400 nM/400 nM, 2: 400 nM/600 nM, and 3: 400 nM/800 nM. (b) M: DL 1000 DNA Marker; 1–4: The reaction temperature was tested at a range of 37°C to 42°C and (c) M: DL 1000 DNA Marker; 1–4: The reaction time was tested at a range of 5 to 30 minutes. RPA, recombinase polymerase isothermal amplification; RSV, respiratory syncytial virus; ADV, respiratory adenovirus.
CRISPR/Cas12a assay optimization
To obtain the optimal CRISPR/Cas detection system, the incorporated concentrations of ssDNA, crRNA-BC with crRNA-E, and Lbcas12a were optimized for the three different ADV genotypes using their respective recombinant plasmids (108 copies/mL). The ssDNA concentration was varied as 400 nM, 800 nM, and 1600 nM, and a NC group (nuclease-free water) was set for each group. For ssDNA, 1600 nM was selected because the fluorescence signal intensity tended to increase with higher ssDNA concentrations (Figure 4). The crRNA-BC and crRNA-E concentrations were varied as 120 nM, 240 nM, and 360 nM. The optimal crRNA incorporation concentrations were found to be 240 nM for the B genotype, 240 nM for the C genotype, and 120 nM for the E genotype (Figure 5). The LbCas12a concentration was varied as 100 nM, 200 nM, and 300 nM. The results suggested that when the LbCas12a incorporation amount increased, the fluorescence signal intensity did not change significantly. Therefore, the LbCas12a concentration was selected as 100 nM (Figure 6).
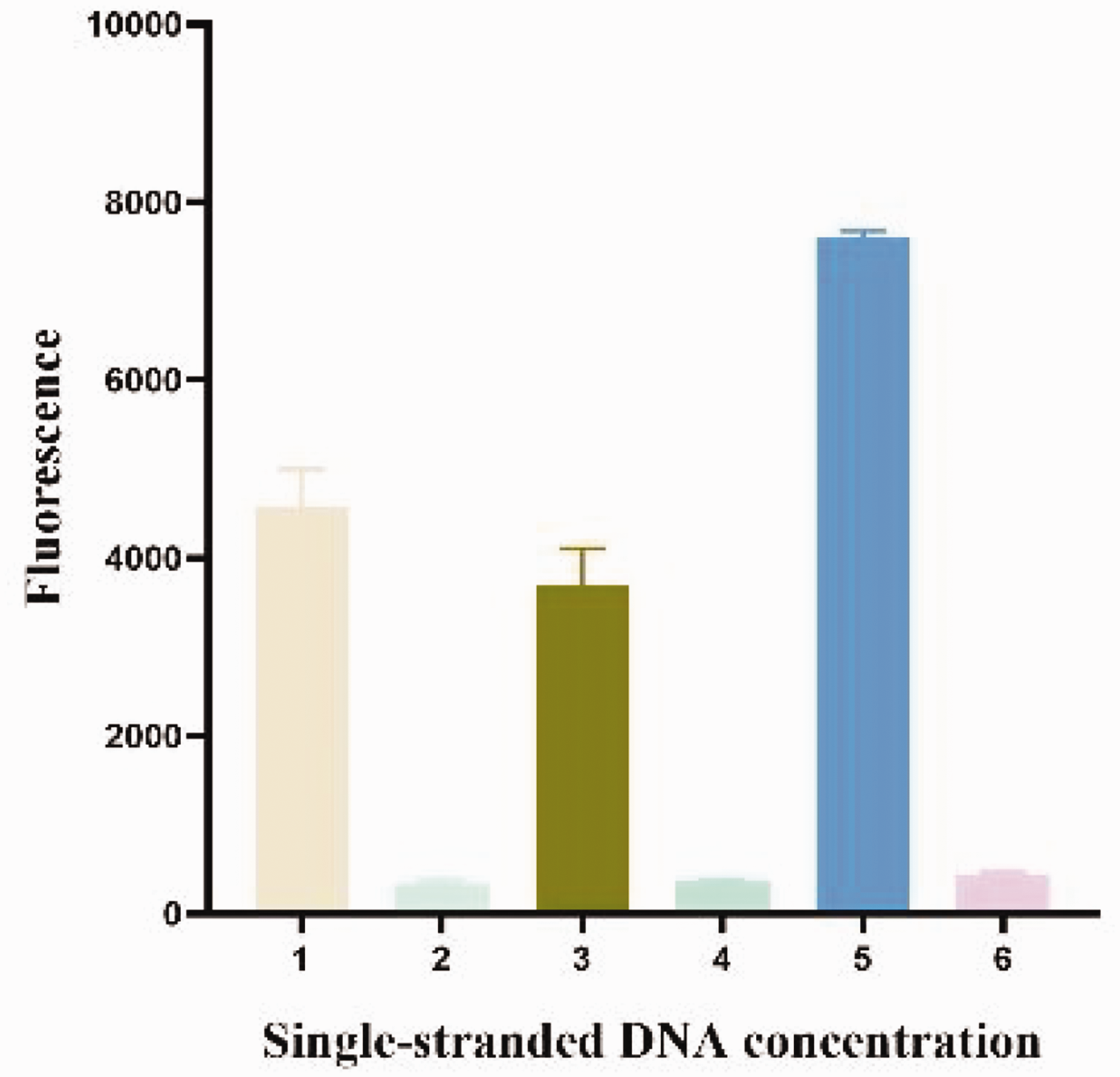
Optimization of the ssDNA concentration in the CRISPR/Cas12a assay. The concentration of ssDNA in the reaction system was tested at 400 nM, 800 nM, and 1600 nM. ssDNA, single-stranded DNA.

Optimization of the crRNA concentrations by CRISPR/Cas12a for three common ADV genotypes. Using recombinant plasmids for B, C, and E ADV as templates, the concentrations of crRNA-BC and crRNA-E in the reaction system were tested at 120 nM, 240 nM, and 360 nM. The negative control group used nuclease-free water as the template. crRNA, CRISPR RNA; ADV, respiratory adenovirus.

Optimization of the LbCas12a concentration in the CRISPR/Cas12a assay. The concentrations of LbCas12a in groups 1, 2, and 3 were 100 nM, 200 nM, and 300 nM, respectively. The corresponding negative control groups used nuclease-free water as the template.
Specificity and sensitivity of the RPA assay
In the RPA step, the RSV-F1/R1 primer pair was used to amplify nucleic acids from pharyngeal swab samples of patients with RSV, ADV, PIV, RhV, MP, CP, FluB, hMPV, FluA, or an RSV/ADV co-infection. The results were negative except for RSV and ADV, which showed specific amplification, indicating that the selected RPA primers did not cross-react with the other eight pathogens examined and were well conserved (Figure 7a). Subsequently, a 10-fold gradient dilution of the RSV and ADV recombinant plasmids was performed using nuclease-free water, with detection limits of 106 copies/mL for both RSV and ADV (Figure 7b). In addition, to verify its reproducibility, the RPA-CRISPR/Cas12a assay was performed using a 10-fold gradient dilution of the plasmids (105 to 107 copies/mL) as the template, then the intra-assay and inter-assay coefficients of variation were analyzed by fluorescence signal intensity. The amplification product bands were analyzed by ImageJ and the gray value data were recorded. The results showed that the intra-assay and inter-assay coefficients of variation were less than 10%, which suggested good reproducibility for this method (Supplementary Table 2).

The specificity and sensitivity values of RPA for RSV and ADV detection. (a) M: DL 1000 DNA marker; The corresponding pathogens 1–10 were RSV, ADV, PIV, RhV, MP, CP, FluB, hMPV, FluA, and nucleic acids from throat swab samples of patients with RSV/ADV co-infection, respectively. 11: Negative control (nuclease-free water) and (b) M: DL 1000 DNA marker; 1–6: 107 to 102 copies/mL of diluted plasmid DNA; 7: Negative control (nuclease-free water). RPA, recombinase polymerase isothermal amplification; RSV, respiratory syncytial virus; ADV, respiratory adenovirus.
Furthermore, our analysis demonstrated that only RSV and ADV each appeared to have fluorescent signal intensities that were significantly different from the control (P < 0.01). The fluorescent signal intensities of the remaining seven pathogens were not statistically different from that of the control (Figure 8).

Specificity analysis of RSV and ADV detected using the RPA-CRISPR/Cas12a assay. (a) Specific detection of RSV and (b) Specific detection of ADV. RSV, respiratory syncytial virus; ADV, respiratory adenovirus; RPA, recombinase polymerase isothermal amplification.
The results indicate that the assay system can specifically detect RSV and ADV with no cross-reactivity with the other seven similar pathogens and is conservative. The detection limits were analyzed using 102 to 107 copies/mL of plasmid DNA of RSV and ADV-B/C/E as templates. For RSV detection, signal from the 104 to 107 copies/mL groups was significantly different from that of the NC group (P = 0.01), while the strongest fluorescence signals of the 103 copies/mL and 102 copies/mL groups were not significantly different from that of the NC group (Figure 9a). For detection of the three ADV genotypes (B, C, and E), signal from the 103 to 107 copies/mL group was significantly different from that of the NC group (P < 0.001 for B and C, P < 0.01 for E), while the strongest fluorescence signal of the 102 copies/mL group was not significantly different from that of the NC group (Figure 9b–d). As a result, detectable RSV RNA concentrations were as low as 104 copies/mL and detectable ADV DNA concentrations were as low as 103 copies/mL.

Sensitivity analysis of RSV and ADV detected using the RPA-CRISPR/Cas12a assay. (a) Sensitivity of RSV detection. (b) Sensitivity of ADV B type detection. (c) Sensitivity of ADV C type detection and (d) Sensitivity of ADV E type detection. RSV, respiratory syncytial virus; ADV, respiratory adenovirus; RPA, recombinase polymerase isothermal amplification.
Specificity and sensitivity of the TaqMan qPCR assay
Nucleic acids from clinical samples of PIV, RhV, MP, CP, FluB, hMPV, and FluA were used as detection templates for the specific detection of RSV and ADV by qPCR. Using RSV and ADV (109 copies/mL) as positive controls and nuclease-free water as negative controls, the results showed that the two qPCR tests were highly specific for detecting RSV or ADV and did not cross-react with similar pathogens (Figure 10). The sensitivity detection experiment was conducted using 103 to 109 copies/mL of RSV or ADV as the template. The CT values of the templates were collated and the lowest detection limit was analyzed (Figure 11). The detection limit for both RSV and ADV was 103 copies/mL. In addition, standard curves were drawn to analyze the accuracy of the results (Supplementary Figures 1 and 2). The R2 values of the standard curves were both greater than 0.99.

Specificity analysis of RSV and ADV detected using TaqMan qPCR. (a) Specific detection of RSV and (b) Specific detection of ADV. RSV, respiratory syncytial virus; ADV, respiratory adenovirus; qPCR, quantitative polymerase chain reaction.
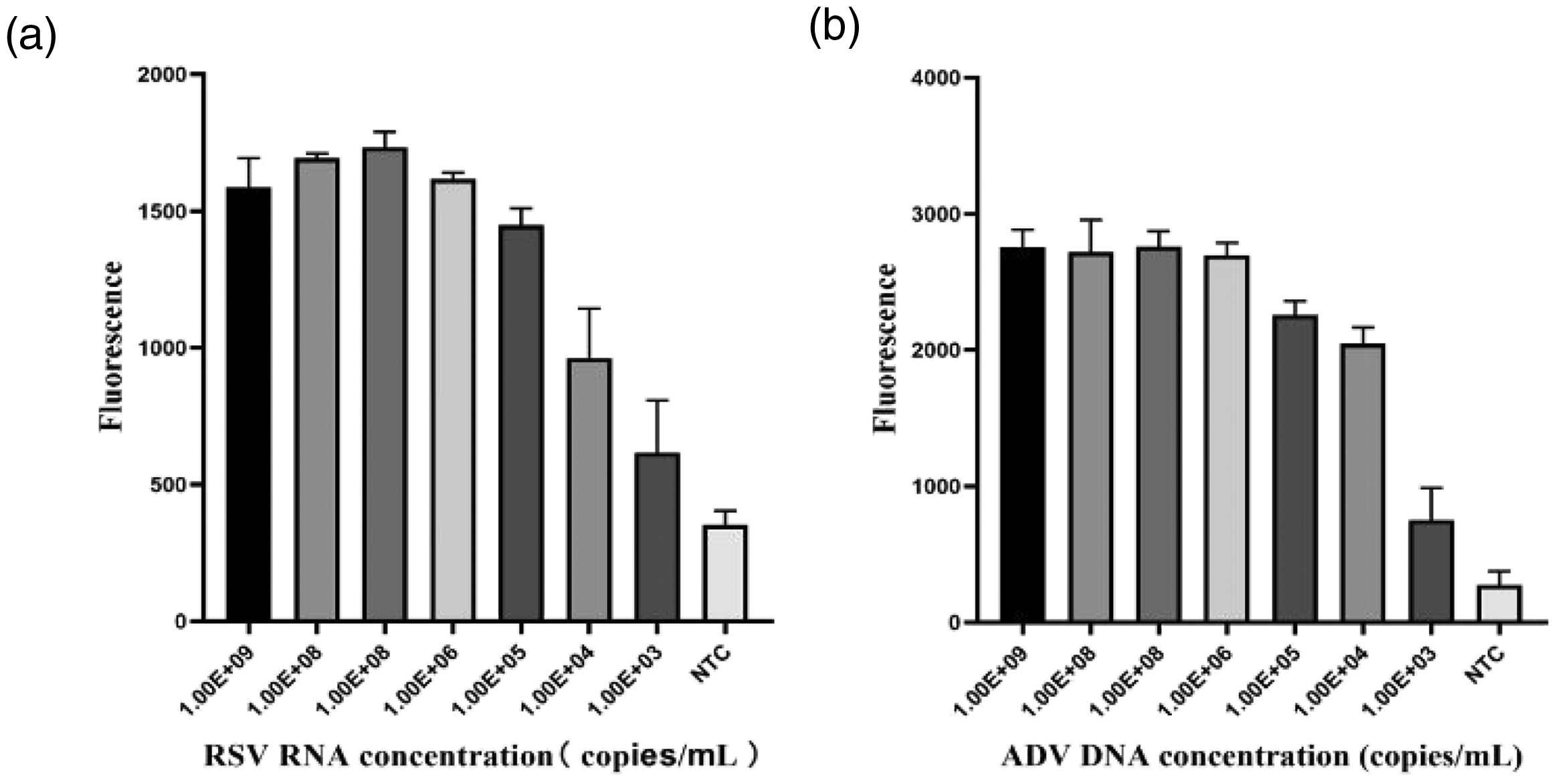
Sensitivity analysis of RSV and ADV detected using TaqMan qPCR. (a) Sensitivity of RSV detection and (b) Sensitivity of ADV detection. RSV, respiratory syncytial virus; ADV, respiratory adenovirus; qPCR, quantitative polymerase chain reaction.
Evaluation of clinical sample diagnostic performance
Pharyngeal swab samples (160) were collected from children with acute respiratory infections. The procedures used for the participants are shown in Figure 12. The qPCR (Supplementary Tables 3 and 4) and RPA-CRISPR (Supplementary Figure 3) methods were simultaneously performed and the results were compared with those of the clinical tests (Table 1).

The procedures for the participants.
Comparison of the RPA-CRISPR/Cas12a and qPCR assays for the detection of clinical throat swab samples

+, positive test result; −, negative test result.
RSV, respiratory syncytial virus; ADV, respiratory adenovirus; qPCR, quantitative polymerase chain reaction; CI, confidence interval.
Compared with the qPCR method, the RPA-CRISPR assay showed high consistency, with Kappa values of 0.988 for RSV and 0.963 for ADV (Supplementary Figure 4). Compared with the qPCR method, we found this new assay to have sensitivity values of 98.7% for RSV detection and 91.4% for ADV detection. The specificity was 100% for both (Figure 12). The RPA-CRISPR method is more suitable for grassroots laboratories, as it involves a shorter detection time, is easy to perform, and can be used to complete experimental operations without relying on a large qPCR instrument. The sensitivity of this method is sufficient for the early detection of disease. The final confirmation of the samples in the experimental operation should be evaluated in conjunction with the actual symptoms of the patients and other test results.
Discussion
With the emergence of the COVID-19 pandemic in 2020, acute respiratory infections have received widespread attention. Because of this, well-performing technologies are needed for the simultaneous detection of multiple pathogens. These methods are required to better differentiate the types of infecting pathogens, accurately detect mixed infections, and achieve rapid and cost-effective access to more pathogen information. 47
In this study, we aimed to develop a dual RPA assay for RSV and ADV, which are common pathogens in acute respiratory tract infections. We combined two specific primer sets in a single tube, then integrated the respective advantages of RPA and CRISPR/Cas technology to establish a rapid detection method. RSV and ADV could be detected at concentrations as low as 104 copies/mL and 103 copies/mL, respectively. Additionally, no cross-reactivity was observed in clinical samples that involved infectious agents that could cause similar symptoms. Compared with the RPA-CRISPR/Cas method used by Gong et al. for RSV type detection, this study has more advantages in terms of detection limit and evaluation of detection performance of clinical samples. Moreover, our method involves not only single RSV detection, but also ADV synchronous detection to preliminarily aim for better assays for multiple pathogen detection. 48 Compared with the multiple reverse transcription-PCR approach conducted by Jiang et al., our method detected comparatively few types of pathogens, but involved relatively simple procedures and has better detection performance. 49
Considering the traditional pathogen detection methods, our assay has several advantages. First, the RPA-CRISPR/Cas12a reaction is performed at a lower temperature that is constant without the need for complex temperature control devices. This can be achieved using a thermostatic heating device in resource-poor regions. Second, the detection time is much lower than those of qPCR methods (>1.5 hours) and the 13 respiratory tests commonly used in laboratories (>3 hours). Third, the simultaneous amplification of two pathogens in a single reaction simplifies the operation steps and shortens the detection time without losing any detection performance. Additionally, the RSV and ADV detection limits were sufficient to reliably detect pathogen-derived nucleic acids in most sample types.
At present, our laboratory has aimed to establish an RPA-CRISPR/Cas12a detection system for three kinds of pathogens, all of which have good detection performance. Considering that the clinical sample verification performed in this study was at a single research center, there may be a certain degree of statistical bias. The number of clinical samples collected can also change with the prevalence rate in the same period of the year. In the future, we will continue to increase the number of samples tested and carry out multi-center studies.
We will also continue to explore multiple detection approaches. In recent years, CRISPR/Cas has received increasing attention from researchers in the molecular diagnostics field because it can be sufficiently compatible with a variety of amplification reactions. This method relies on the specific recognition property of the crRNA to generate detection results with higher accuracy. In addition, CRISPR/Cas can be accessed by a variety of result-reading methods, including, but not limited to, large and small fluorescent detection devices, test strips, and visual interpretation. 50 Although the test strips and some visualization can be more convenient in resource-poor areas, they involve a certain degree of subjectivity. The fluorescence quantitative detection method is more credible, but may potentially be inconvenient in some cases. In the future, we will develop RPA multiplexing, optimization, and lyophilization of the basic components and attempt to combine the CRISPR/Cas portion of the assay with a microfluidic detection device to truly maximize its accuracy.
Conclusion
We established a synchronous detection platform for RSV and ADV that uses both RPA and CRISPR/Cas12a detection technologies. This method has the advantages of high sensitivity, high specificity and simple operation, with certain clinical application value that is suitable for immediate detection.
Supplemental Material
sj-pdf-1-imr-10.1177_03000605231223083 - Supplemental material for A multiplex recombinase polymerase amplification assay combined with CRISPR/Cas12a for the detection of respiratory syncytial virus and respiratory adenovirus
Supplemental material, sj-pdf-1-imr-10.1177_03000605231223083 for A multiplex recombinase polymerase amplification assay combined with CRISPR/Cas12a for the detection of respiratory syncytial virus and respiratory adenovirus by Gao Hongdan, Du Yao, Chai Qiang, Huang Meng, Liu Xiaorong, Xing Zhihao and Ma Dongli in Journal of International Medical Research
Footnotes
Acknowledgements
We thank the Clinical Laboratory of Shenzhen Hospital for providing the pharyngeal swab samples and the Pathogen High-throughput sequencing engineering laboratory team of Shenzhen Children’s Hospital for their support.
Author contributions
GH designed and performed the experiments, conducted statistical analysis, and wrote a draft of the manuscript. DY assisted with sample acquisition, writing the manuscript, and performing the experiments. XZ, CQ, and HM conceptualized the study, performed the epidemiological investigation, designed the primers, and performed the bioinformatics analysis. LX conceptualized the study and revised the manuscript. MD revised the manuscript, supervised the study, and is responsible for project administration, financial support, and correspondence. All authors read and approved the final version of the manuscript.
Data availability statement
De-identified data are available from the corresponding author upon request.
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Funding
The project was funded by the Promotion of Pathogen High-Throughput Gene Sequencing Technology Engineering Laboratory of Shenzhen (Project No. Deepafai (2019) 986) and the Research platform for Mechanism and Diagnosis of Pathogens of Respiratory Tract in Children (Project No. SEY-GSP-YXPT-A02).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.