
Abstract
Hypophysitis is an extremely rare inflammatory disease that can mimic the clinical and radiological features of a pituitary adenoma. In this case report, we describe a 45-year-old woman with secondary xanthogranulomatous hypophysitis (XGH) who presented with signs of a pituitary macroadenoma. The patient complained of headaches, visual impairment, and amenorrhea-galactorrhea syndrome. Her physical examination was normal. Laboratory investigation revealed corticotropin, thyrotropin, and gonadotropin deficiencies. She also had low visual acuity in her right eye and an altered visual field. Pituitary magnetic resonance imaging revealed an intra and suprasellar mass measuring 13 × 11 × 16 mm, with hemorrhagic necrosis, that was having a discrete mass effect on the patient’s optic chiasm and pituitary stalk. The patient was treated with hydrocortisone and levothyroxine, and then transferred to the Neurosurgery department for total transsphenoidal resection of the mass. Histological examination of the tumor permitted a diagnosis of XGH of a remodeled Rathke’s pouch cyst to be made. Systemic conditions such as tuberculosis, sarcoidosis, and other granulomatous diseases were excluded. The etiopathogenesis of XGH remains poorly characterized, but it may be a progressive form of lymphocytic hypophysitis or a remodeled Rathke’s pouch cyst. Screening for autoimmune pathology and systemic diseases is essential to guide appropriate management.
Introduction
Hypophysitis is a rare inflammatory disorder of the pituitary gland that can be classified as primary or secondary, depending on its underlying cause. Primary hypophysitis, also known as idiopathic hypophysitis, is caused by an autoimmune or unknown inflammatory or infiltrative process that only affects the pituitary gland.1,2 Secondary hypophysitis is caused by systemic granulomatous diseases, infections, sellar tumors or cysts, or drugs.1,2 Pituitary hypophysitis can be further categorized histologically as lymphocytic, granulomatous, xanthomatous, necrotizing, and xanthogranulomatous hypophysitis (XGH). 3 The last of these is a rare subtype that consists of a combination of xanthomatous and granulomatous lesions. However, its pathogenesis is not well understood. Hypophysitis can cause pituitary enlargement and hypopituitarism, and therefore it can be confused with pituitary adenoma, owing to their similar clinical and radiological features. 4
In this report, we present a case of secondary XGH in which the patient initially presented with features of a pituitary macroadenoma.
Case presentation
A 45-year-old woman was admitted to the Department of Endocrinology of our hospital in May 2022 for the treatment of a pituitary macroadenoma. She had a history of type 2 diabetes, hypertension, coronary artery disease, and euthyroid multinodular goiter, but no family history of autoimmune disease. Three years previously, she had presented with headache, visual impairment, and amenorrhea-galactorrhea syndrome. Initial investigations revealed mild hyperprolactinemia (72.55 ng/mL). Pituitary magnetic resonance imaging (MRI) examination revealed a pituitary macroadenoma of 15.5-mm diameter, a thin pituitary stalk that was displaced to the right, and the absence of the spontaneous hypersignal of the posterior pituitary lobe. The patient was treated with cabergoline 0.5 mg weekly but was lost to follow-up until May 2022, when she was referred to the Department of Endocrinology because of a recurrence of the same symptoms. On physical examination, she was found to have a body mass of 76 kg and a height of 167 cm, corresponding to a body mass index of 27.2 kg/m2, a blood pressure of 130/80 mmHg, a heart rate of 69 beats/minute, and a multinodular goiter. However, the physical examination was otherwise unremarkable. Further investigations revealed corticotropin, thyrotropin, and gonadotropin deficiencies (Table 1). An ocular examination revealed low visual acuity (4/10 for the right eye), an altered visual field, and right eye predominance. There was no papillary edema. Pituitary MRI examination revealed an intra and suprasellar mass measuring 13 × 11 × 16 mm that included hemorrhagic necrosis and had a discrete mass effect on her optic chiasm and pituitary stalk (Figure 1). These findings suggested a diagnosis of pituitary macroadenoma associated with apoplexy. The patient was treated with hydrocortisone and levothyroxine, and then transferred to the Neurosurgery department for total transsphenoidal resection of the mass. The surgeons described the mass as either a craniopharyngioma or a remodeled Rathke’s pouch cyst (RPC). Histological examination revealed necrotic debris containing lanceolate cholesterol crystals with a polymorphic granuloma in contact with the necrotic tissue. It was composed of lymphocytes, plasma cells, and multinucleated macrophagic giant cells, as well as foamy histiocytes with clear cytoplasm (Figures 2 and 3). Postoperatively, the patient’s symptoms improved, but her hypopituitarism persisted and she developed diabetes insipidus. Therefore, she was treated with hydrocortisone, levothyroxine, and desmopressin.
Key laboratory data prior to surgery

HbA1c, glycosylated hemoglobin; ACTH, adrenocorticotropic hormone; FT4, free thyroxine; TSH, thyroid-stimulating hormone; FSH, follicle-stimulating hormone; LH, luteinizing hormone.
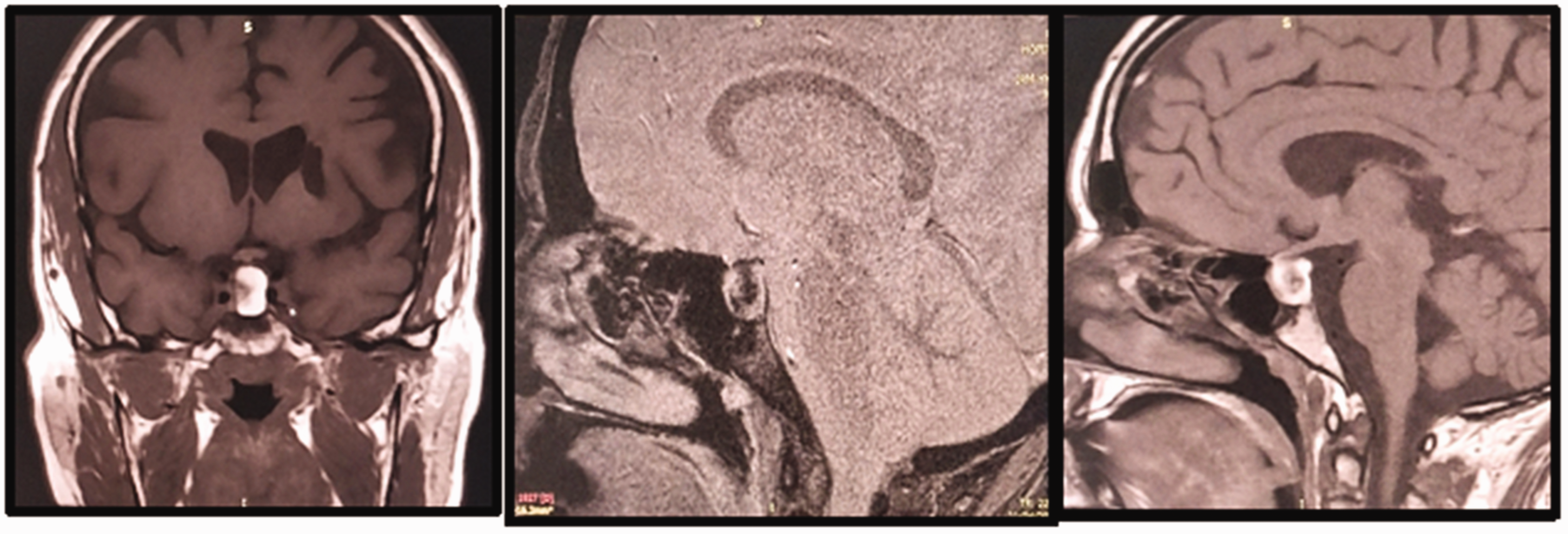
Pituitary magnetic resonance images of the patient. a) Image showing enlargement of the sella turcica, with a well-defined left-sided lesion measuring 13 × 11 × 16 mm. The tumor was having a mass effect on the optic chiasm and pituitary stalk. It was also in contact with the left internal carotid artery, which remained permeable and of normal diameter. The pituitary stalk was thin and displaced to the right. b) The lesion extended to the suprasellar space, was rounded, and showed a heterogenous hypersignal on T1-weighted examination. It did not show enhancement following the injection of gadolinium, but showed punctuate anomalies, owing to intrasellar bleeding. The tumor extended to the roof of the sphenoid sinus and c) The normal hypersignal of the posterior hypophysis was not visible.
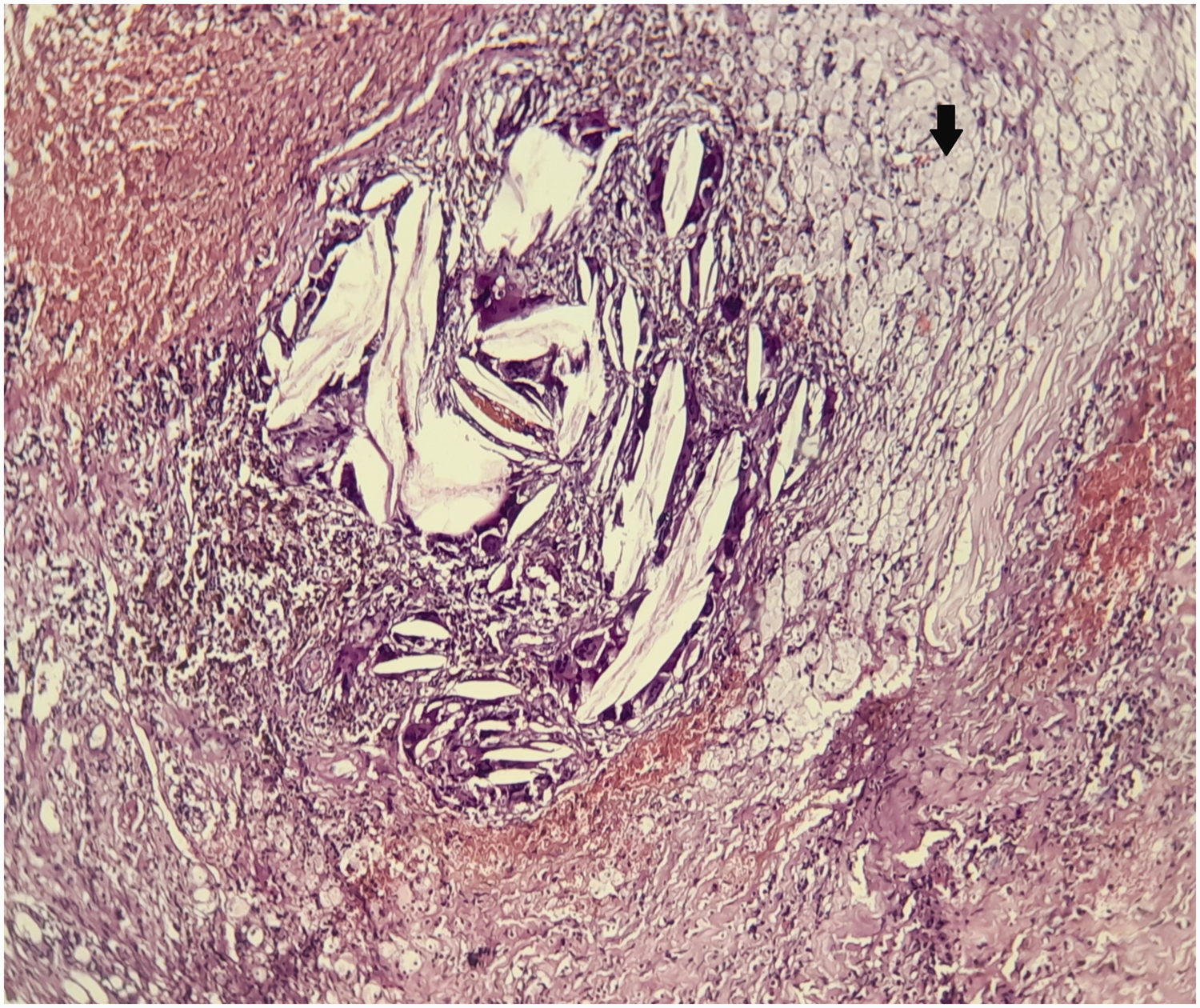
Hematoxylin and eosin-stained section through the patient’s tumor. Magnification ×20. Xanthogranuloma, with a polymorphous inflammatory infiltrate composed of lymphocytes and plasma cells, as well as numerous xanthoma cells (arrow).

Hematoxylin and eosin-stained section through the patient’s tumor. Magnification ×40. Foreign body-like multinucleate giant cells (arrow) in contact with cholesterol clefts (star) are visible.
Tuberculosis was suspected as the etiology of the condition, but this was ruled out after a series of negative test results (chest X-ray, tuberculosis skin test, and sputum and urinary culture for Koch’s bacillus). Furthermore, the patients did not have adenomegaly, fever, respiratory symptoms, knotty erythema, or uveitis; and her angiotensin converting enzyme activity was normal, at 43 IU/L (normal range, 35 to 70 IU/L). Computed tomography and bone scintigraphy examinations did not reveal any lesions. A salivary gland biopsy was histologically normal. Therefore, a diagnosis of XGH of a remodeled RPC was made through the exclusion of granulomatous diseases as possible diagnoses.
The reporting of this case conforms to the CARE guidelines. 5 Written informed consent was obtained from the patient for her treatment and for the publication of this case report. Ethics approval for this case report was not required because of the retrospective nature of the study.
Discussion
Xanthomatous hypophysitis (XH) is a rare subtype of secondary hypophysitis that was first described by Folkerth in 1998. 6 It affects both men and women, and usually develops at approximately 40 years of age. The lesions associated can range from XH to XGH to xanthogranuloma (XG), which have differing clinicopathological characteristics. 7 XGH is an extremely rare form of pituitary hypophysitis that may present with similar clinical and radiological features to other pituitary lesions, such as pituitary adenoma, as in the present case.
The present patient presented with headache, visual impairment, and amenorrhea-galactorrhea syndrome secondary to disconnection hyperprolactinemia. The most commonly described presenting symptoms of granulomatous hypophysitis are headache, reported by 61% of patients, followed by visual disturbances, reported by 41.5% of patients, and polyuria-polydipsia syndrome, reported by 26.8% of patients. 8 The association between infundibular enlargement on imaging and central diabetes insipidus has been described by many authors.9,10
The most commonly described laboratory abnormalities are corticotropin deficiency, present in 73.1% of patients, growth hormone deficiency, present in 66.7% of patients, and gonadotropin deficiency, present in 66% of patients. 8 The present patient had hypopituitarism, with corticotropin, thyrotropin, and gonadotropin deficiencies, which were the result of inflammatory infiltration of the pituitary gland.
MRI is the best imaging methodology for the characterization of pituitary lesions. Hypophysitis can present with various imaging features. Symmetrical enlargement of the pituitary and thickening of the pituitary stalk represent the most common features. According to Zhu et al., 11 most patients with XH have cystic sellar lesions characterized by peripheral enhancement on MRI. However, this can also be a feature of other subtypes of hypophysitis. 12
A definitive diagnosis of XGH is made histologically, in the presence of foamy histiocytes, multinucleated giant cells containing cholesterol clefts, and lymphocytes. However, the variation in the histological findings, which can be both cystic and infiltrative (lymphocytes, foamy histiocytes, and lipid-rich lymphocytes and histiocytes), raises a question regarding its true nature. According to McKeel et al., 13 it may represent a spectrum of disease, with purely lymphocytic hypophysitis being the predominant early lesion, and the granulomatous component representing a later phase of the disease process. XH is histologically characterized by the presence of foamy histiocytes and lymphoplasmacytic infiltrates within the pituitary gland or sellar region, while XGH typically contains substantial hemosiderin deposits, in addition to cholesterol clefts, lymphoplasmacytic infiltrates, fibrosis, multinucleated giant cells, eosinophilic necrotic debris, and macrophage accumulation. Given that hemosiderin accumulates in XGH but not in XH, it has been suggested that XH can progress to XGH through repeated episodes of hemorrhage, rupture, or leakage of the RPC. 7
Pituitary imaging of the present patient revealed the presence of intrasellar bleeding, and macroscopic examination revealed the liquid component to be a remodeled RPC, which was consistent with an adamantinomatous craniopharyngioma, as discussed above. The histological classification of craniopharyngioma as adamantinomatous or papillary is well established and clinically relevant. 14 Xanthogranulomatous changes, consisting of cholesterol clefts; chronic inflammatory infiltration with macrophages and/or multinucleated giant cells; necrotic debris; hemosiderin deposits; and fibrosis are usually considered to be characteristic of the adamantinomatous variant. 15 Accordingly, it has been proposed that the presence of a suprasellar mass consisting of xanthogranulomatous tissue suggests a diagnosis of adamantinomatous craniopharyngioma, even in the absence of epithelium. 16 This form is characterized by suprasellar extension and the presence of calcification, and is rarely associated with pituitary insufficiency. 15 In the present patient, no calcification was identified on pituitary imaging or pathological examination.
The pathological mechanisms underpinning autoimmune pituitary disorders merit some discussion. The present patient did not have a history of autoimmune disorders, but her sex and age may suggest the progression of LH to XGH by the time of presentation. However, infection or systemic disease may also have been involved in the etiology. Systemic conditions such as tuberculosis, sarcoidosis, and other granulomatous diseases were excluded as etiologic factors in the present patients.
Surgical resection is typically the only treatment that is required. 17 However, anti-inflammatory and immunosuppressive drugs may be indicated in patients with residual or recurrent hypophysitis.17,18
Conclusion
In the present report, we have described a patient with a rare pituitary pathology who presented with hypopituitarism and visual impairment and was misdiagnosed as having a macroadenoma. Postoperative histological evaluation revealed an XGH of a remodeled RPC. Following surgical treatment, the patient had persistent pituitary dysfunction. Surgical intervention early in the development of such lesions may have beneficial effects on pituitary function, because chronic inflammation leads to the destruction of the pituitary gland and permanent pituitary dysfunction.
Footnotes
Acknowledgements
We would like to express our deepest gratitude to Professor Hafedh Jemel and our colleagues from the Department of Radiology of La Rabta University Hospital for their invaluable assistance in managing the patient.
Author Contributions
IO: conception and design; acquisition, analysis, and interpretation of data; manuscript drafting. SS: acquisition, analysis, and interpretation of data; manuscript drafting. YM, AZ, NK, EK, MY, and MC: acquisition, analysis, and interpretation of data. MC: critical revision of the manuscript for important intellectual content. All the authors were involved in the management of the patient and the revision of the manuscript, and approved the submitted version of the manuscript.
Ethics statement
Ethics approval for this case report was not required, because of the retrospective nature of the study. The written informed consent of the patient was obtained for the publication of this report.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.